1. 领域背景与文献引入
文献英文标题:A potential role of the JNK pathway in hyperoxia-induced cell death, myofibroblast transdifferentiation and TGF-β1-mediated injury in the developing murine lung;发表期刊:BMC Cell Biology;影响因子:未公开;研究领域:新生儿支气管肺发育不良(BPD)发病机制与信号通路调控
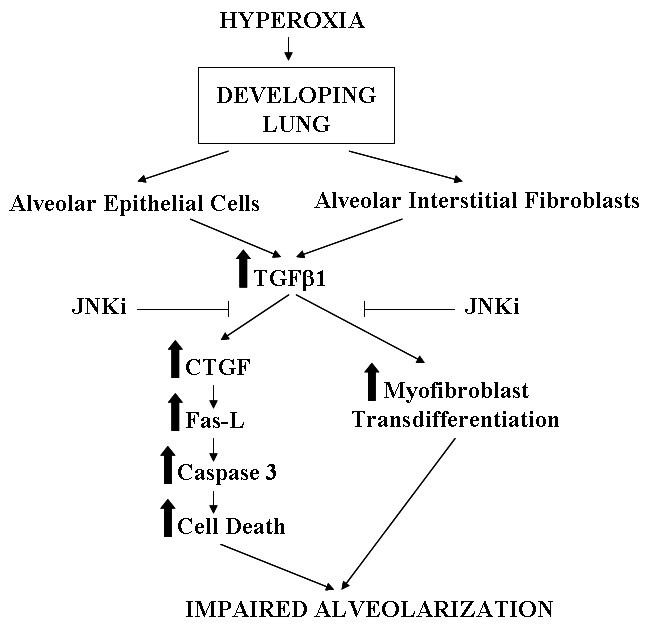
支气管肺发育不良是早产儿最常见的慢性肺部疾病,高氧暴露是诱导该病发生的关键环境因素之一。随着无创通气技术在早产儿中的普及,高氧对发育肺的损伤作用愈发受到关注,但目前BPD的发病率仍呈上升趋势,核心原因在于对高氧诱导肺损伤的分子机制理解不足,缺乏精准的治疗靶点。领域共识:丝裂原活化蛋白激酶(MAPK)家族中的c-Jun氨基末端激酶(JNK)通路参与调控细胞增殖、分化、死亡及炎症反应,转化生长因子-β1(TGF-β1)已被证实参与高氧诱导的肺细胞死亡与肺泡化障碍,但JNK通路与TGF-β1信号在发育肺损伤中的协同调控机制尚未完全明确。针对这一研究空白,本研究系统探讨了JNK通路在高氧诱导的肺上皮细胞死亡、肺成纤维细胞转分化及TGF-β1介导的肺泡化障碍中的作用,为BPD的机制研究和治疗靶点开发提供了关键依据。
2. 文献综述解析
本文献综述部分以“分子通路-细胞响应-疾病表型”为核心分类维度,系统梳理了领域内现有研究。现有研究已证实高氧暴露可诱导发育肺损伤并最终导致BPD,TGF-β1是介导高氧诱导细胞死亡和肺泡化障碍的关键细胞因子,JNK通路可调控细胞增殖、死亡及炎症反应,且部分研究提示JNK通路与TGF-β1信号存在关联。已有的体外细胞模型和体内转基因模型为研究肺发育损伤提供了有效的技术支撑,但现有研究存在明显局限性:多数研究聚焦单一细胞类型或单一通路的独立作用,未系统探讨JNK通路在高氧诱导的多种肺损伤效应中的调控作用,也未明确JNK与TGF-β1在发育肺损伤中的协同调控机制,导致对BPD发病机制的理解存在碎片化问题。本研究的创新价值在于,首次通过体外细胞模型和体内动物模型的联合验证,系统证实JNK通路是高氧诱导的上皮细胞死亡、肌成纤维细胞转分化及TGF-β1介导肺泡化障碍的核心调控节点,同时明确了JNK抑制剂对高氧诱导肺损伤的治疗潜力,填补了通路协同调控机制的研究空白。
3. 研究思路总结与详细解析
本研究的核心目标是明确JNK通路在高氧诱导的发育肺损伤中的调控作用,核心科学问题为JNK通路是否介导高氧诱导的细胞死亡、肌成纤维细胞转分化及TGF-β1介导的肺损伤,技术路线遵循“体外细胞模型验证通路依赖性→体内转基因模型验证治疗效果→整合调控机制”的闭环逻辑。
3.1 高氧诱导肺上皮细胞死亡的JNK通路依赖性验证
实验目的:明确高氧诱导的肺上皮细胞死亡是否依赖JNK通路的激活。方法细节:使用人肺腺癌A549细胞和小鼠肺上皮MLE-12细胞,分别暴露于21%(常氧)、40%、60%、95%氧浓度环境中24h或48h,部分细胞提前1h用5μM JNK抑制剂SP600125预处理;通过台盼蓝排斥实验、TUNEL实验检测细胞活力与凋亡水平,免疫印迹(Western Blot)检测JNK磷酸化水平及细胞死亡标志物FAS配体(FAS-L)、活化半胱天冬酶3(caspase 3)的蛋白表达。结果解读:高氧以剂量依赖方式降低细胞活力,95%氧浓度处理24h后A549细胞活力降至常氧组的42%(n=4,P<0.01),同时JNK磷酸化水平显著升高;JNK抑制剂可使各氧浓度组的细胞活力恢复至常氧组的90%以上(n=4,P<0.01),并显著抑制FAS-L和活化caspase 3的蛋白表达(FAS-L表达下调68%,n=4,P<0.02),证实高氧诱导的肺上皮细胞死亡依赖JNK通路的激活。
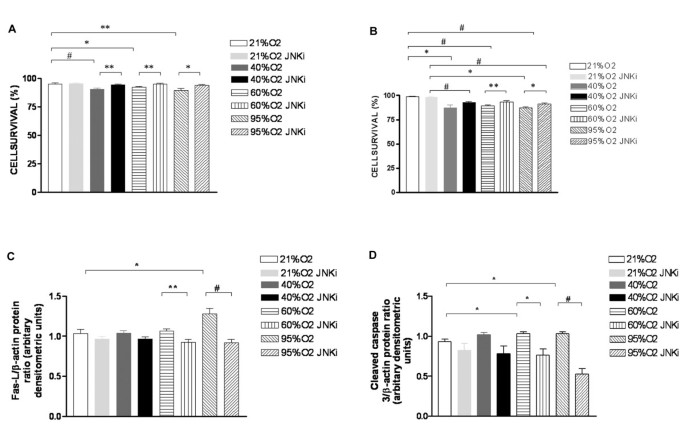
实验所用关键产品:JNK抑制剂SP600125(Calbiochem)、TUNEL试剂盒(Roche Diagnostics)、JNK及磷酸化JNK抗体(Cell Signaling Technology、Promega)、FAS-L、caspase 3抗体(Cell Signaling Technology、Santa Cruz Biotechnology)。
3.2 高氧诱导肺成纤维细胞转分化的JNK通路依赖性验证
实验目的:验证高氧诱导的肺泡间质成纤维细胞(AIF)向肌成纤维细胞(MYF)转分化是否依赖JNK通路。方法细节:分离胎鼠AIF细胞,暴露于95%氧浓度环境24h,部分细胞提前1h用1μM CEP-1347或10μM SP600125预处理;通过油红O染色检测AIF标志物脂滴的表达,免疫荧光检测MYF标志物α-平滑肌肌动蛋白(α-SMA)的表达,免疫印迹检测过氧化物酶体增殖物激活受体γ(PPARγ)、脂肪细胞分化相关蛋白(ADRP)、纤连蛋白、LEF-1的蛋白水平。结果解读:高氧处理后AIF的脂滴染色完全消失,α-SMA荧光强度升高3.2倍(n=4,P<0.01),同时PPARγ和ADRP水平分别下调72%和65%,纤连蛋白和LEF-1水平分别上调2.1倍和1.8倍(n=4,P<0.05);JNK抑制剂可完全逆转上述标志物的表达变化,证实AIF向MYF的转分化过程依赖JNK通路的激活。
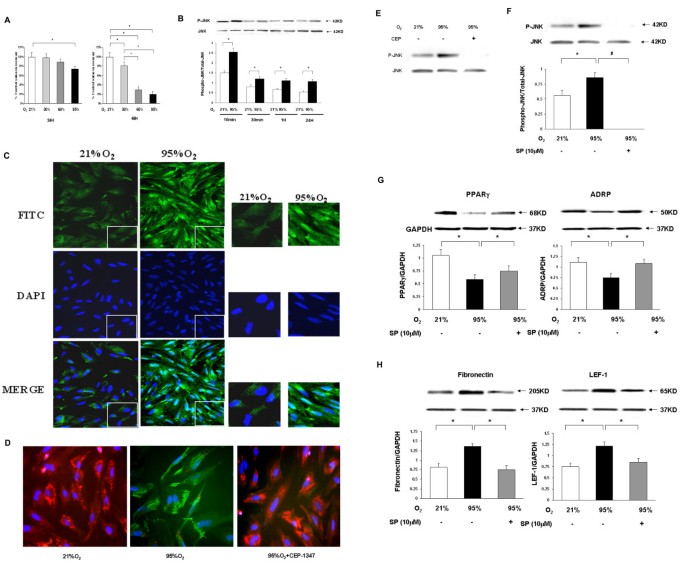
实验所用关键产品:JNK抑制剂CEP-1347(Cephalon馈赠)、SP600125(Calbiochem)、PPARγ抗体(Santa Cruz Biotechnology)、ADRP抗体(Constantine Londos馈赠)。
3.3 高氧对TGF-β1及CTGF表达的调控作用验证
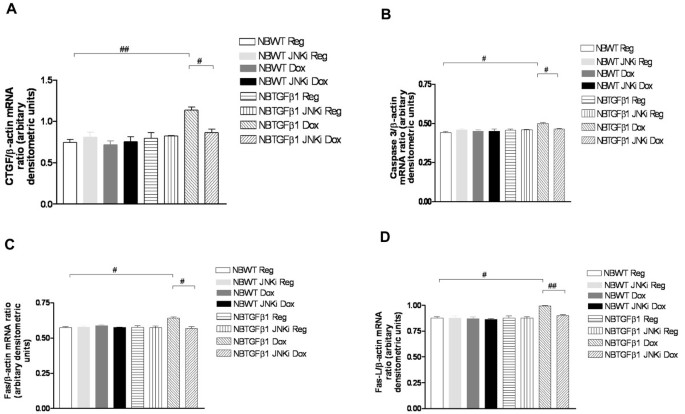
实验目的:明确高氧是否诱导肺上皮细胞中TGF-β1和结缔组织生长因子(CTGF)的表达上调。方法细节:将A549细胞暴露于21%、40%、60%、95%氧浓度环境24h,通过半定量RT-PCR检测TGF-β1和CTGF的mRNA表达水平。结果解读:高氧以剂量依赖方式上调TGF-β1和CTGF的mRNA表达,95%氧浓度处理后TGF-β1 mRNA水平升高2.7倍,CTGF mRNA水平升高3.1倍(n=4,P<0.01),证实高氧可激活TGF-β1-CTGF信号轴。
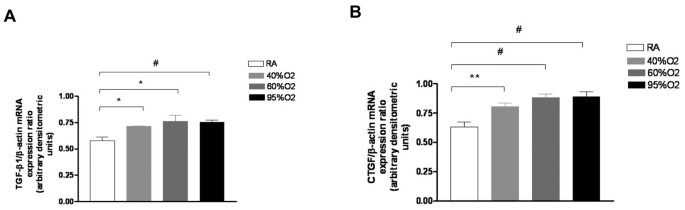
实验所用关键产品:TRIzol试剂(Invitrogen)、定制化RT-PCR引物。
3.4 体内JNK通路抑制对高氧诱导肺损伤的治疗效果验证
实验目的:验证JNK抑制剂在体内模型中对高氧诱导的死亡率和肺泡化障碍的改善作用。方法细节:使用新生TGF-β1转基因(TG)小鼠和野生型(WT)小鼠,分为常氧/高氧(100%氧)组,部分小鼠每日腹腔注射20mg/kg的SP600125,观察出生后1-7天(PN1-PN7)的存活率;对PN7-PN10的TGF-β1 TG小鼠注射JNK抑制剂,通过肺组织苏木精-伊红(HE)染色、形态计量学分析肺泡化程度,RT-PCR检测细胞死亡标志物和CTGF的mRNA表达,ELISA检测支气管肺泡灌洗液(BALF)中TGF-β1的蛋白水平。结果解读:高氧暴露下TGF-β1 TG小鼠的7天存活率仅为32%,显著低于WT小鼠的68%(n=4,P<0.001);JNK抑制剂可使WT小鼠存活率提升至100%(P<0.0001),TG小鼠存活率提升至76%(P=0.01)。在常氧环境下,TGF-β1 TG小鼠存在明显的肺泡化障碍,肺泡弦长较WT小鼠增加45%(n=3,P<0.01);JNK抑制剂可使肺泡弦长降低18%(n=3,P<0.01),同时显著抑制caspase 3、FAS、FAS-L及CTGF的mRNA表达(下调幅度均超过50%,n=3,P<0.01),且不影响BALF中TGF-β1的蛋白水平,证实JNK通路抑制通过调控TGF-β1下游信号发挥作用。
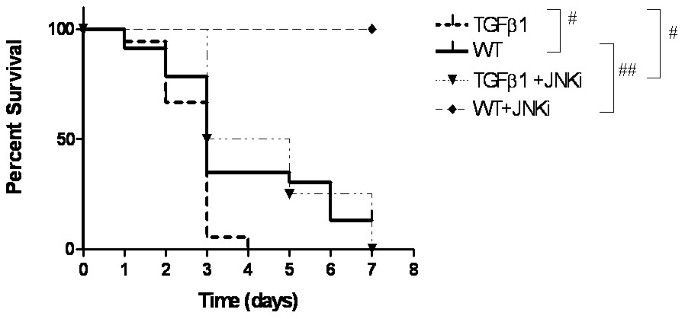
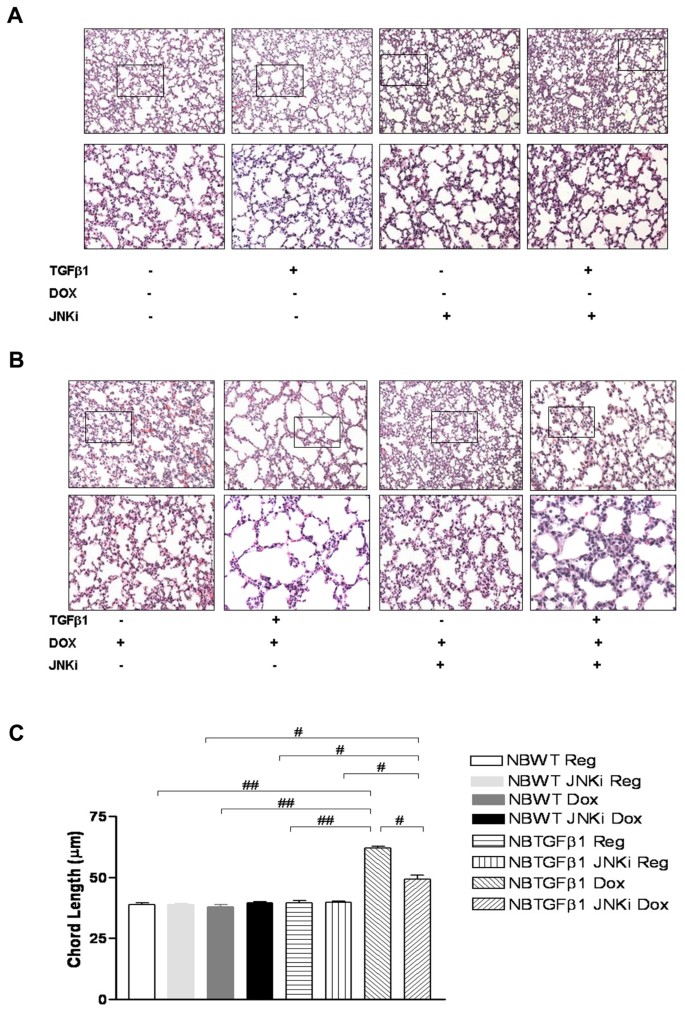
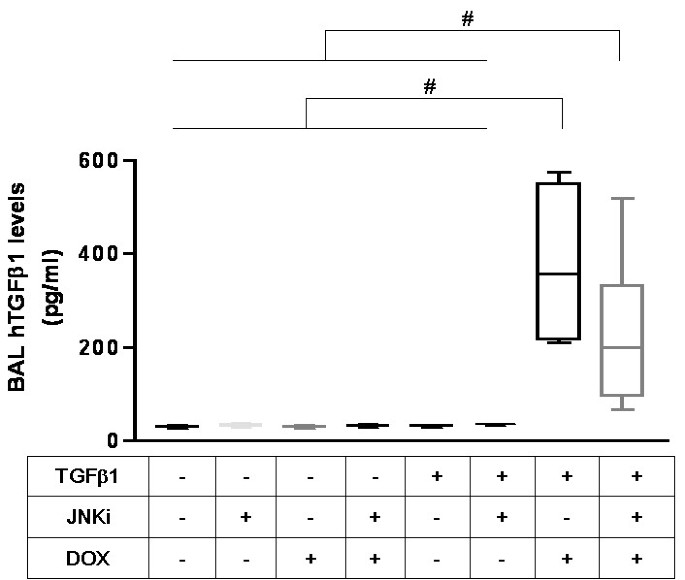
实验所用关键产品:JNK抑制剂SP600125、TGF-β1 ELISA试剂盒(R&D Systems)、HE染色试剂。
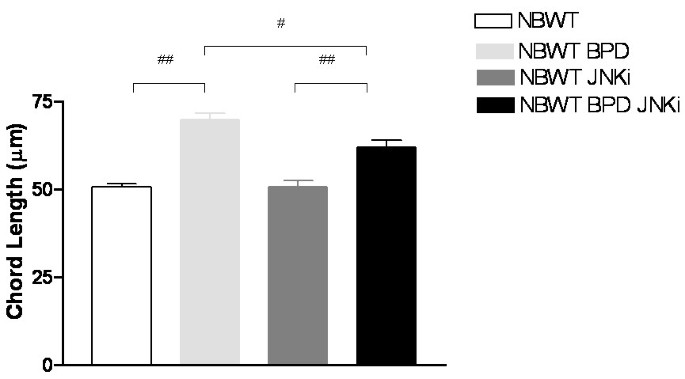
3.5 野生型BPD模型中JNK通路抑制的效果验证
实验目的:验证JNK抑制剂在经典BPD模型中的治疗潜力。方法细节:将新生WT小鼠暴露于100%氧环境中PN1-PN4,随后恢复常氧至PN14,部分小鼠每日注射JNK抑制剂,通过肺形态计量学分析肺泡化程度。结果解读:BPD模型小鼠的肺泡弦长较常氧组增加38%(n=4,P<0.001),JNK抑制剂可使肺泡弦长降低22%(n=4,P<0.01),显著改善肺泡化障碍,证实JNK通路是BPD的潜在治疗靶点。
4. Biomarker研究及发现成果解析
Biomarker定位与筛选逻辑
本研究涉及的Biomarker包括三类:通路激活标志物(磷酸化JNK)、细胞功能标志物(细胞死亡标志物FAS-L、活化caspase 3;肌成纤维细胞转分化标志物α-SMA、PPARγ)、疾病相关细胞因子(TGF-β1、CTGF)。筛选与验证逻辑为:基于高氧诱导肺损伤的已知通路,先在体外细胞模型中筛选标志物的表达变化及与JNK通路的关联,再通过体内动物模型验证其与疾病表型的相关性及治疗响应性。
研究过程详述
Biomarker来源涵盖体外细胞裂解液、细胞培养上清及体内肺组织、BALF样本。验证方法包括:免疫印迹检测蛋白表达水平、免疫荧光检测细胞标志物定位、半定量RT-PCR检测mRNA表达、ELISA检测细胞因子蛋白浓度。特异性与敏感性数据显示:高氧诱导A549细胞中磷酸化JNK水平升高2.4倍(n=4,P<0.01),FAS-L蛋白表达上调2.5倍(n=4,P<0.02);JNK抑制剂可使高氧处理的A549细胞活力恢复至常氧水平的92%(n=4,P<0.01);在体内模型中,JNK抑制剂使TGF-β1 TG小鼠的肺泡弦长降低18%(n=3,P<0.01),同时使CTGF mRNA表达下调56%(n=3,P<0.01)。
核心成果提炼
本研究证实磷酸化JNK可作为高氧诱导肺损伤的通路激活标志物,其表达水平与细胞死亡、肌成纤维细胞转分化程度呈正相关;TGF-β1和CTGF可作为高氧诱导肺损伤的效应标志物,JNK通路是其下游关键调控节点;JNK抑制剂可显著改善高氧诱导的小鼠死亡率和肺泡化障碍,提示JNK通路可作为BPD的潜在治疗靶点。本研究的创新性在于首次系统证实JNK通路在高氧诱导的多种肺损伤效应中的核心调控作用,为BPD的机制研究和治疗靶点开发提供了关键的Biomarker和理论依据。