1. 领域背景与文献引入
文献英文标题:Tissue of origin determines cancer-associated CpG island promoter hypermethylation patterns;发表期刊:Genome Biology;影响因子:未公开;研究领域:肿瘤表观遗传学(CpG岛启动子高甲基化与肿瘤起源组织关联性研究)
肿瘤表观遗传学是肿瘤研究的核心领域之一,CpG岛启动子异常高甲基化是肿瘤中最常见的表观遗传重编程事件之一。1999年Toyota等首次提出结直肠癌的CpG岛甲基化表型(CIMP),后续研究在乳腺癌、胶质母细胞瘤等多种肿瘤中发现类似表型,传统观点认为该事件通过沉默抑癌基因直接驱动肿瘤进展,这一模型在2000年代初成为领域主流认知。然而,随着研究深入,领域内逐渐出现争议:部分研究发现高甲基化的基因在正常起源组织中已处于沉默状态,如RUNX3在胃癌中频繁高甲基化,但在正常胃上皮细胞中从未表达,提示高甲基化可能并非直接导致基因沉默的驱动事件。同时,现有研究多聚焦单一肿瘤类型,缺乏跨多肿瘤类型的系统性分析,无法明确不同肿瘤中高甲基化模式差异的核心决定因素,这一空白限制了对肿瘤表观遗传重编程共性机制的理解。本研究针对这一核心问题,通过分析7种不同组织来源的1154例肿瘤甲基化谱,系统性探究肿瘤起源组织对高甲基化模式的调控作用,为重新认识肿瘤高甲基化的生物学意义提供依据。
2. 文献综述解析
作者按“传统模型争议-单一肿瘤研究局限-前期研究延伸”的逻辑梳理领域研究现状,明确现有研究的核心分歧与空白。
现有研究可分为两类:一类支持传统“高甲基化驱动抑癌基因沉默”模型,这类研究通过甲基化特异性PCR、亚硫酸氢盐测序等技术,在多种肿瘤中验证了BRCA1、APC等抑癌基因的高甲基化现象,技术优势在于能精准检测单个基因的甲基化状态,为抑癌基因失活机制提供了表观遗传学证据,但局限性在于多聚焦单个基因或单一肿瘤类型,缺乏跨肿瘤的系统性分析,无法解释不同肿瘤中高甲基化模式的差异;另一类研究对传统模型提出质疑,发现高甲基化的基因在正常组织中已处于沉默状态,如RUNX3在胃癌中的高甲基化并未导致基因沉默,因为该基因在正常胃上皮中本就不表达,这类研究挑战了“高甲基化直接驱动肿瘤进展”的认知,但未明确不同肿瘤中高甲基化模式差异的核心调控因素。作者前期在乳腺癌中的研究发现,高甲基化基因具有谱系特异性,在正常起源组织中已被沉默,这一结果为本次跨7种肿瘤类型的系统性研究奠定了基础,也明确了本研究的创新方向:填补跨肿瘤类型高甲基化模式调控机制的研究空白,明确肿瘤起源组织对高甲基化模式的决定作用。
与现有研究相比,本研究的创新价值在于首次通过大样本跨肿瘤分析,明确肿瘤起源组织的基因表达模式是高甲基化模式的核心决定因素,突破了传统模型的认知局限,提出高甲基化更多是伴随肿瘤发生的表观遗传“乘客”事件,而非直接驱动肿瘤进展的“驱动”事件,为领域重新认识肿瘤表观遗传重编程的生物学意义提供了系统性证据。
3. 研究思路总结与详细解析
本研究的核心目标是明确不同肿瘤类型中CpG岛启动子高甲基化模式的决定因素,核心科学问题为“肿瘤起源组织的哪些分子特征调控了高甲基化基因的选择?”,技术路线遵循“多组学数据整合分析→基因特征筛选→关联性验证→机制模型提出”的闭环逻辑,通过系统性分析7种肿瘤的甲基化与基因表达数据,逐步揭示肿瘤起源组织对高甲基化模式的调控机制。
3.1 多肿瘤甲基化数据整合与高甲基化基因集定义
实验目的:构建跨7种肿瘤类型的高甲基化易感与抵抗基因集,明确不同肿瘤中高甲基化模式的差异特征。
方法细节:收集乳腺、结直肠、前列腺、肺、卵巢、急性髓系白血病、胶质母细胞瘤7种肿瘤共1149例样本的Illumina Infinium HumanMethylation27 BeadChip甲基化数据,统一采用β值(甲基化信号强度与总信号强度的比值)衡量甲基化水平,定义“高甲基化易感基因”为在对应正常组织中未甲基化(β<0.3)、且在≥20%肿瘤样本中甲基化(β>0.3)的基因,“高甲基化抵抗基因”为在所有肿瘤样本中均未甲基化的基因;同时,通过与3项独立乳腺癌研究的高甲基化基因集对比,验证方法的可重复性。
结果解读:共鉴定出1009个高甲基化易感基因,其中约一半(503个)为单一肿瘤类型特有,无基因在所有7种肿瘤中均发生高甲基化;主成分分析显示,前3个主成分可将7种肿瘤样本完全区分(图1c),提示肿瘤类型是高甲基化模式差异的核心决定因素;乳腺癌高甲基化基因集与3项独立研究的重叠率达81.5%(n=316,P<2×10^-16),验证了研究方法的可靠性。
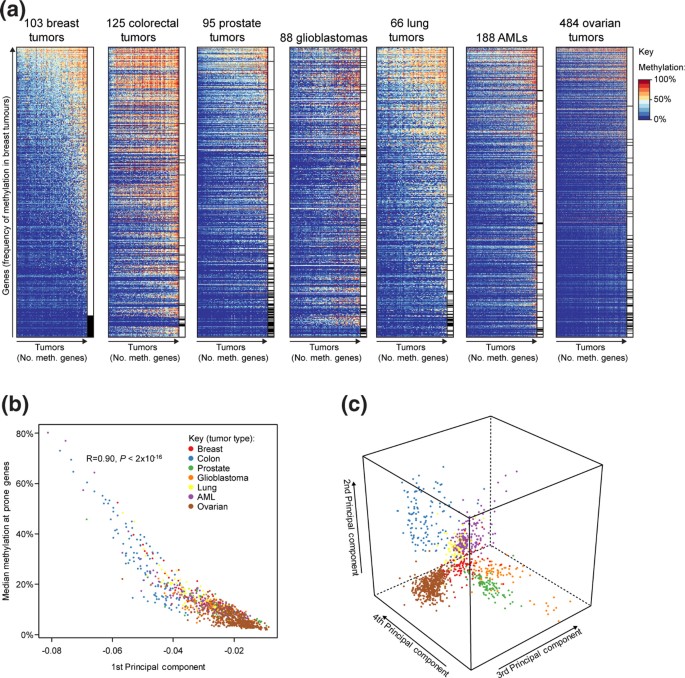
产品关联:实验所用关键产品:Illumina Infinium HumanMethylation27 BeadChip甲基化芯片。
3.2 高甲基化易感基因的分子特征分析
实验目的:解析高甲基化易感基因与抵抗基因的功能、表达及序列特征差异,筛选潜在调控因素。
方法细节:通过Ensembl Biomart进行基因本体论(GO)富集分析,比较两类基因的功能差异;采用基于信息论的方法量化基因在正常组织中的特异性表达水平;从UCSC基因组浏览器下载重复元件注释信息,分析转录起始位点(TSS)周围±2kb范围内LINE、SINE、LTR三类重复元件的密度;通过PhastCons跨物种序列比对分析启动子区域的进化保守性;检测基因邻近长链基因间非编码RNA(lincRNA)的富集情况。
结果解读:GO富集分析显示,高甲基化抵抗基因显著富集于管家功能相关术语(如“有丝分裂细胞周期”“RNA加工”),而易感基因显著富集于发育相关术语(如“系统发育”“器官发育”)(图2a);易感基因的组织特异性表达评分显著高于抵抗基因(Wilcoxon秩和检验,P<0.001)(图2b);易感基因TSS周围的重复元件密度显著低于抵抗基因(Fisher精确检验,P<0.001)(图3a);易感基因启动子区域的进化保守性显著高于抵抗基因(Wilcoxon秩和检验,P<0.001)(图3b);易感基因邻近lincRNA的比例显著高于抵抗基因(Fisher精确检验,P<0.05)(图3d),提示易感基因具有组织特异性表达、启动子序列保守性高、重复元件少的特征。
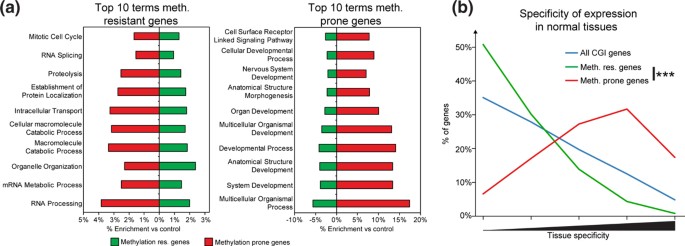
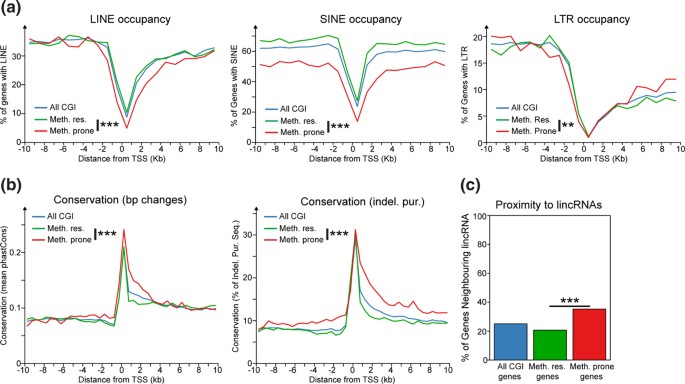
产品关联:文献未提及具体实验产品,领域常规使用GO富集分析工具(如DAVID、Ensembl Biomart)、跨物种保守性分析工具(如PhastCons)、R语言及Bioconductor包进行数据分析。
3.3 肿瘤起源组织基因表达与高甲基化的关联性验证
实验目的:验证肿瘤起源组织的基因表达状态是高甲基化易感的核心决定因素。
方法细节:定义“可变甲基化(VM)基因”为在至少一种肿瘤类型中从未发生高甲基化的易感基因,“一致甲基化(CM)基因”为在所有7种肿瘤类型中均发生高甲基化的易感基因;分析VM基因在不同肿瘤起源正常组织中的表达水平,比较结直肠癌中正常结肠与肝脏组织差异表达基因的甲基化水平差异。
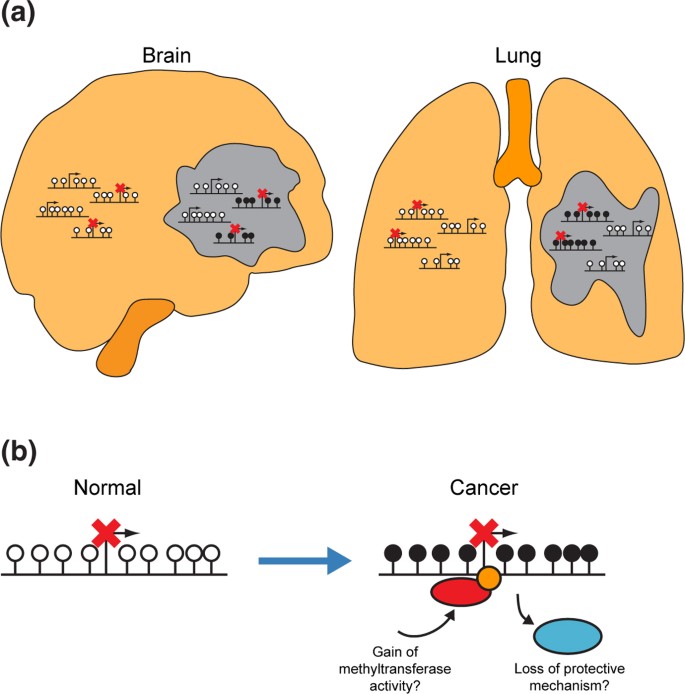
结果解读:VM基因在其未发生高甲基化的肿瘤起源正常组织中呈高表达状态,如PAX6在胶质母细胞瘤中未发生高甲基化,且在正常脑组织中持续高表达;在结直肠癌中,正常结肠组织中沉默的基因在肿瘤中的甲基化水平显著高于正常结肠中激活的基因(单侧Wilcoxon秩和检验,P=1.6×10^-7)(图4d);这一结果明确提示,肿瘤起源组织中沉默的基因更易发生高甲基化,起源组织的基因表达模式直接决定了高甲基化的基因选择。
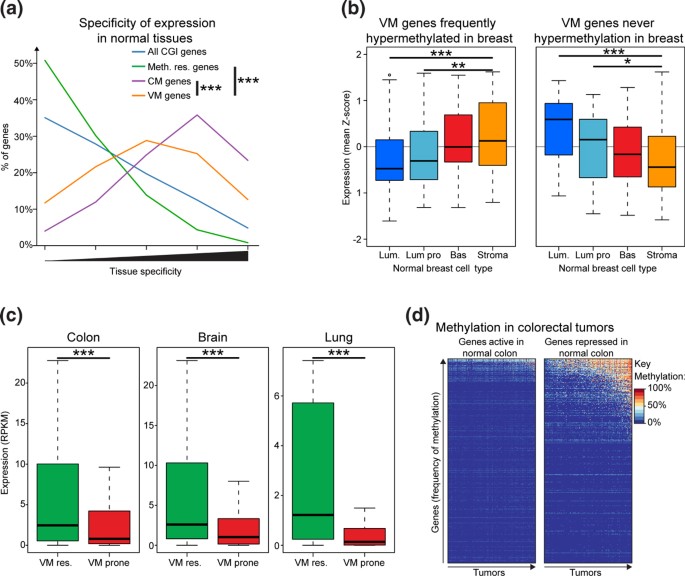
产品关联:文献未提及具体实验产品,领域常规使用基因表达数据分析工具(如R语言、Bioconductor包)进行差异表达分析与甲基化关联分析。
3.4 机制模型提出与临床关联性验证
实验目的:基于上述结果提出肿瘤高甲基化的调控模型,并与临床数据整合验证模型的合理性。
方法细节:整合高甲基化基因特征、起源组织表达关联及领域内相关研究,提出“肿瘤起源组织中沉默的基因通过表观遗传调控机制紊乱发生高甲基化”的模型;分析已知抑癌基因的高甲基化频率,对比具有CpG岛甲基化表型(CIMP)的肿瘤与临床预后的关联性。
结果解读:已知抑癌基因(如BRCA1、APC)的高甲基化频率远低于起源组织沉默基因的高甲基化频率,如BRCA1仅在12%的卵巢癌和2%的乳腺癌中发生高甲基化,提示高甲基化多为伴随肿瘤发生的“乘客”事件;同时,具有CIMP表型的肿瘤(如结直肠癌、乳腺癌、胶质母细胞瘤)预后更好,这与“高甲基化驱动肿瘤进展”的传统模型矛盾,进一步支持本研究提出的模型:高甲基化是表观遗传调控机制紊乱的伴随事件,而非直接驱动肿瘤进展的因素(图5)。
产品关联:文献未提及具体实验产品,领域常规使用Kaplan-Meier分析等工具进行临床预后关联性分析。
4. Biomarker研究及发现成果解析
本研究鉴定的核心Biomarker为“肿瘤起源组织特异性高甲基化基因集”,属于表观遗传特征类Biomarker,其筛选与验证逻辑为“多肿瘤甲基化数据整合定义易感基因→正常组织表达数据验证组织特异性→跨肿瘤类型验证关联性”,形成完整的证据链。
该Biomarker的来源为7种不同组织来源的肿瘤甲基化谱及对应正常组织的基因表达数据,验证方法包括主成分分析(区分不同肿瘤类型)、Wilcoxon秩和检验(比较基因表达与甲基化的关联性)、Fisher精确检验(分析基因特征差异);特异性方面,主成分分析显示该基因集可通过甲基化模式准确区分7种不同组织来源的肿瘤,前3个主成分的方差解释率达10.4%,敏感性方面,单一肿瘤类型特有高甲基化基因占总易感基因的50%,提示对肿瘤起源组织具有较高的特异性与敏感性。
核心成果方面,该Biomarker的功能关联在于揭示了肿瘤表观遗传重编程的组织特异性调控机制,创新性在于首次明确肿瘤起源组织的基因表达模式是高甲基化模式的核心决定因素,突破了传统“高甲基化直接驱动抑癌基因沉默”的认知,提出高甲基化多为伴随肿瘤发生的“乘客”事件;统计学结果显示,乳腺癌高甲基化基因集与独立研究的重叠率达81.5%(n=316,P<2×10^-16),结直肠癌中正常沉默基因与激活基因的甲基化水平差异显著(P=1.6×10^-7),为Biomarker的可靠性提供了严谨的统计学支持。此外,该Biomarker为肿瘤起源组织的表观遗传学鉴定提供了潜在的分子标签,也为重新评估高甲基化基因的抑癌基因属性提供了依据,提示不能仅依据基因在肿瘤中的高甲基化状态就认定其为抑癌基因,需结合正常组织中的表达状态进行判断。