1. 领域背景与文献
文献英文标题:Resolving sampling and population-size biases in domestication genomics supports a South Asian origin of walnuts;发表期刊:Genome Biology;影响因子:17.906(2023年JCR);研究领域:植物驯化基因组学、群体遗传学。
领域共识:植物驯化基因组学是解析作物起源、扩散和人工选择机制的核心研究领域,2000年群体结构分析软件(STRUCTURE)的发布为群体遗传结构解析提供了核心工具,2009年混合祖先分析软件(ADMIXTURE)的推出进一步提升了大样本聚类的运算效率,随着高通量测序成本的下降,近10年该领域已解析了水稻、玉米、小麦等主要作物的驯化历史。当前研究热点包括多年生作物驯化机制解析、古基因组学在起源研究中的应用、群体聚类方法的偏倚校正等。该领域尚未解决的核心问题包括:群体聚类算法普遍受抽样不平衡和有效种群大小差异的影响,易将大的祖先种群错误判定为混合种群;多年生木本作物由于世代周期长、种群结构复杂,其驯化起源研究普遍存在方法学偏倚校正缺失的问题。
普通核桃是全球重要的木本油料作物,其驯化起源地长期存在争议,现有假说包括西亚伊朗-安纳托利亚起源、中亚天山起源、南亚喜马拉雅区域起源三类,此前的研究受限于地理抽样不平衡(南亚样本量不足)和方法学偏倚,未能形成统一结论。本研究针对上述研究空白,首先优化群体结构分析软件的参数策略,同时校正抽样不平衡和有效种群大小差异导致的偏倚,进而明确普通核桃的驯化起源地,解析其扩散路径,筛选驯化相关候选基因,为核桃遗传改良提供理论基础,同时为作物驯化基因组学研究提供方法学参考。
2. 文献综述解析
核心信息段:作者的综述按“方法学挑战→研究对象争议”的二维逻辑展开,系统梳理了群体聚类方法的偏倚来源和核桃起源研究的现有进展。
现有研究中,支持性结论包括:群体结构分析软件和混合祖先分析软件是群体遗传结构解析的主流工具,抽样不平衡会导致聚类结果出现错误,将抽样量少的群体错误合并,已有研究提出调整祖先先验参数和较小的初始ALPHA值可以缓解抽样不平衡导致的偏倚;核桃起源的三类假说均有对应的考古学和遗传学证据支持,比如西亚地区发现了2300年前的核桃花粉记录,中亚地区存在大片野生核桃林,南亚克什米尔地区发现了4700-4000年前的核桃壳遗存。现有技术的优势包括:群体结构分析软件的参数可调性优于混合祖先分析软件,其内置的相关等位基因频率模型(F模型)理论上可以处理不同种群间的有效种群大小差异,有效校正遗传漂移导致的偏倚。现有研究的局限性包括:此前的方法优化仅能单独校正抽样不平衡或有效种群大小差异其中一种偏倚,尚未有研究整合两种校正策略;核桃起源研究普遍存在南亚样本量不足的问题,且未考虑不同地理种群的有效种群大小差异,导致聚类结果将祖先种群判定为混合种群,进而得出错误的起源地结论。
本研究的创新价值在于:首次提出整合相关等位基因频率模型、替代祖先先验和更小初始ALPHA值的参数策略,可同时校正两种偏倚,准确识别祖先种群;首次基于多维度群体基因组学证据明确支持普通核桃起源于南亚,解决了长达数十年的起源争议;筛选得到一批调控壳厚度、油脂含量等关键驯化性状的候选基因,为核桃分子育种提供了靶点。推测:此前支持核桃西亚或中亚起源的研究,其结论很可能受到抽样不平衡和有效种群大小差异偏倚的影响,导致群体聚类结果出现错误。
3. 研究思路总结与详细解析
核心信息段:本研究的整体目标为建立可同时校正抽样和种群大小偏倚的群体聚类方法,明确普通核桃的驯化起源地,筛选驯化相关候选基因;核心科学问题包括如何同时消除群体结构分析中的两类偏倚,以及普通核桃的真实驯化起源地;技术路线遵循“方法优化→模拟验证→核桃群体基因组分析→多证据交叉验证起源地→选择清除分析筛选驯化基因”的闭环逻辑,结果可靠性高。
3.1 群体结构分析软件参数优化与模拟验证
实验目的:开发可同时校正抽样不平衡和有效种群大小差异偏倚的群体结构分析软件参数策略,通过模拟验证其准确性。
方法细节:设置两组参数方案,ParamSet1采用替代祖先先验(POPALPHA=1)、初始ALPHA值设为0.25,同时结合相关等位基因频率模型;ParamSet2采用相同的祖先先验和ALPHA值,结合非相关等位基因频率模型(即已有研究提出的仅校正抽样偏倚的策略)。使用溯祖模拟软件模拟符合作物驯化场景的三群体数据集,包含1个大的祖先种群和2个经历瓶颈的衍生种群,样本量和单核苷酸多态性数量与实际核桃数据集匹配,分别用两组参数进行群体结构聚类分析。
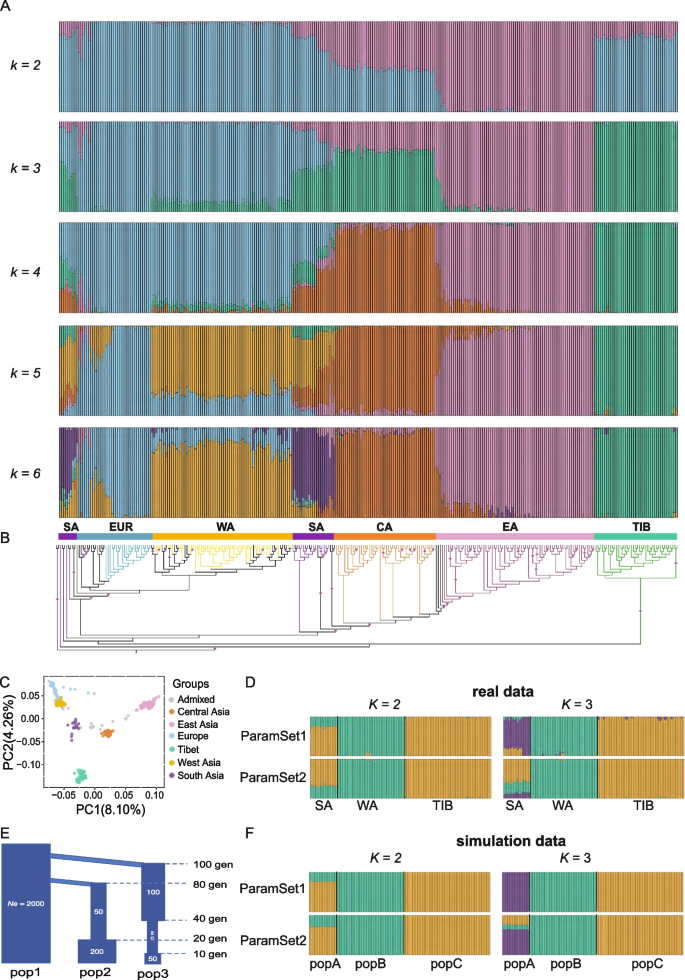
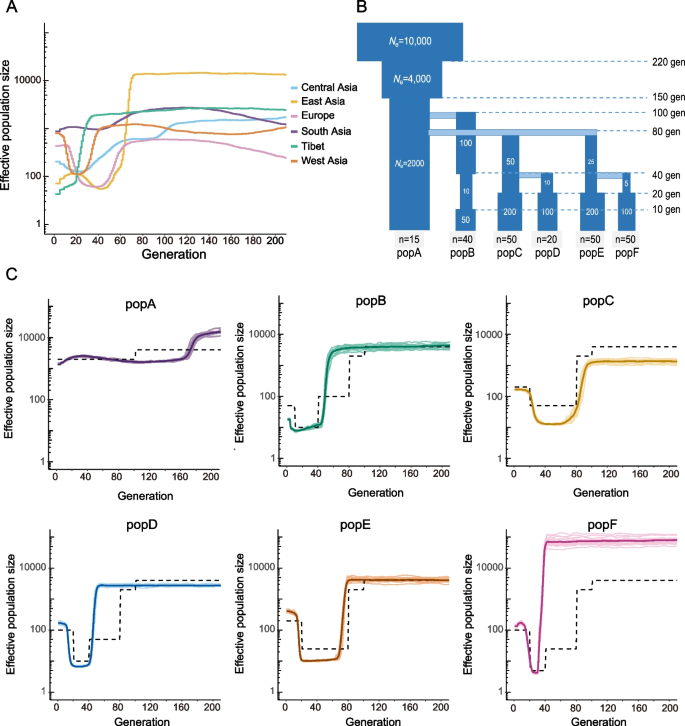
结果解读:模拟数据的分析结果显示,ParamSet1可以正确识别出3个独立遗传聚类,而ParamSet2仅能识别出2个聚类,且错误地将祖先种群判定为两个衍生种群的混合(对应文献图1F)。该结果证实ParamSet1可同时校正两类偏倚,准确识别驯化过程中的祖先种群。
产品关联:文献未提及具体实验产品,领域常规使用群体结构分析软件、溯祖模拟软件等群体遗传学分析工具。
3.2 核桃群体遗传结构分析
实验目的:解析全球普通核桃的群体遗传结构,识别祖先种群。
方法细节:整合本研究新测序和已发表的共298份普通核桃种质的全基因组重测序数据,样本覆盖东亚、中亚、欧洲、西亚、西藏、南亚等全部分布区域,经过序列质量控制、参考基因组比对、变异检测后,筛选得到14950个独立的非编码单核苷酸多态性位点。分别用ParamSet1和ParamSet2进行群体结构聚类,结合邻接进化树、主成分分析验证聚类结果。
结果解读:ParamSet1下最优聚类数为K=6,可将样本分为东亚、中亚、欧洲、西亚、西藏、南亚6个独立遗传群组,其中南亚种群的遗传漂移参数Fk值最低,为0.0565,说明其经历的遗传漂移最弱,保留的祖先遗传变异最多(对应文献图1A、1B、1C)。而ParamSet2下最优聚类数为K=5,无法将南亚种群识别为独立群组,错误判定其为混合种群,与模拟结果一致。
产品关联:文献未提及具体实验产品,领域常规使用高通量测序平台、序列比对软件、变异检测软件、进化分析软件等工具。
3.3 种群遗传多样性与人口动态历史分析
实验目的:从遗传多样性、突变负荷、人口动态历史等维度,验证南亚种群的祖先地位。
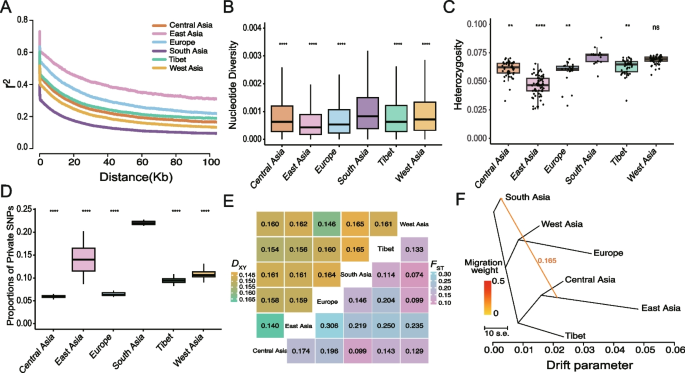
方法细节:计算6个地理群组的连锁不平衡衰减、核苷酸多样性、观测杂合度、私有单核苷酸多态性比例、遗传分化系数和绝对遗传距离,使用混合图分析软件推断群体间的基因流事件;使用连锁分析软件计算基于纯合子片段的近交系数,使用变异功能注释软件预测有害突变和功能缺失突变,比较不同群组的突变负荷;使用有效种群大小推断软件分析不同种群的有效种群大小动态变化,结合模拟验证瓶颈事件对有效种群大小推断的影响。
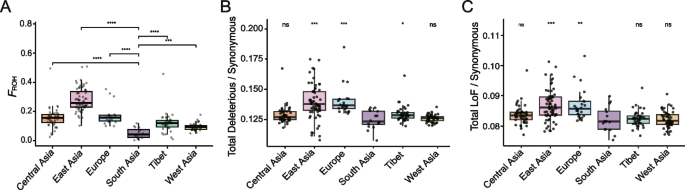
结果解读:南亚种群的连锁不平衡衰减最快,核苷酸多样性最高,观测杂合度最高,校正样本量后的私有单核苷酸多态性比例为21.4%−22.7%(n=16,P<0.0001,与其余群体比较),为所有群组最高(对应文献图2A-2D);基于纯合子片段的近交系数最低,有害突变/同义突变比值、功能缺失突变/同义突变比值均为所有群组最低(n=16,P<0.001,与其余群体比较,对应文献图3A-3C);人口动态历史分析显示,南亚种群的有效种群大小长期保持稳定,没有经历明显的瓶颈事件,而其他衍生种群均经历了不同程度的瓶颈(对应文献图4A)。上述所有遗传特征均符合起源地种群的预期。
产品关联:文献未提及具体实验产品,领域常规使用群体遗传多样性分析软件、变异功能注释软件、有效种群大小推断软件等工具。
3.4 叶绿体基因组单倍型分析
实验目的:从细胞质遗传角度验证南亚作为核桃起源地的地位。
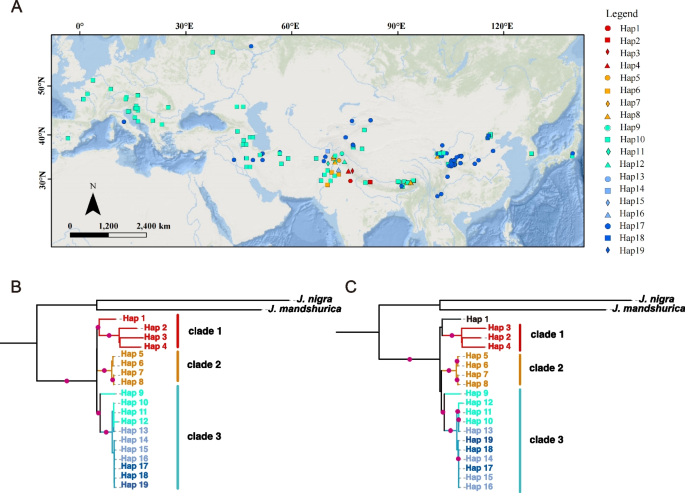
方法细节:组装298份样本的叶绿体基因组,结合已发表的7份喜马拉雅地区核桃的叶绿体基因组数据,鉴定叶绿体单倍型,构建最大似然进化树和邻接进化树,分析单倍型的地理分布模式。
结果解读:总共鉴定到19个叶绿体单倍型,其中南亚种群拥有14个单倍型,包括12个区域特有单倍型,单倍型多样性为所有区域最高;进化树显示南亚的单倍型位于拓扑结构的基部节点,支持其祖先地位(对应文献图5A-5C)。
产品关联:文献未提及具体实验产品,领域常规使用序列比对软件、进化树构建软件、单倍型分析软件等工具。
3.5 驯化相关候选基因筛选
实验目的:筛选核桃驯化过程中受到正向选择的关键基因。
方法细节:以南亚种群为参考群体,使用跨群体扩展单倍型纯合性分析方法,对5个衍生种群(东亚、中亚、欧洲、西亚、西藏)进行选择清除分析,筛选在所有衍生种群中均存在一致等位基因频率偏移的位点,对位点所在或临近的基因进行功能注释。
结果解读:共鉴定到45个受到正向选择的候选基因,其中29个有明确功能注释,包括参与细胞壁重塑的FEI2基因(调控坚果壳结构)、参与花粉管生长的果胶酯酶基因(调控育性)、参与脂质转运的ABCG7基因(调控种仁油脂含量),这些基因对应核桃驯化的关键目标性状。另有未注释基因JreChr03G10963存在两个非同义单核苷酸多态性位点,在南亚种群中的频率为0.5937和0.7187(n=16),在所有衍生种群中接近固定,可能是调控核桃驯化性状的关键未解析基因。
产品关联:文献未提及具体实验产品,领域常规使用选择清除分析软件、基因功能注释工具等。
4. Biomarker 研究及发现成果
核心信息段:本研究涉及的Biomarker为“南亚普通核桃种群的特异性遗传特征组合”,属于群体遗传类起源诊断标志物,其筛选验证逻辑为“优化参数的群体结构聚类识别→核基因组遗传多样性验证→人口动态历史验证→叶绿体基因组单倍型验证”的多证据交叉验证链条。
研究过程详述:该Biomarker的检测样本为全球范围内298份普通核桃的叶片基因组DNA,覆盖所有主要分布区域。验证方法包括全基因组重测序、单核苷酸多态性基因分型、群体遗传参数计算、叶绿体基因组组装、单倍型分析等。其中,南亚种群的私有单核苷酸多态性比例为21.4%−22.7%(n=16,P<0.0001,与其余群体比较),核苷酸多样性较东亚群体高41%(n=16,P<0.0001),有害突变/同义突变比值较东亚群体低32%(n=16,P<0.001),叶绿体单倍型数量占所有已鉴定单倍型的73.7%,特异性和辨识度极高。
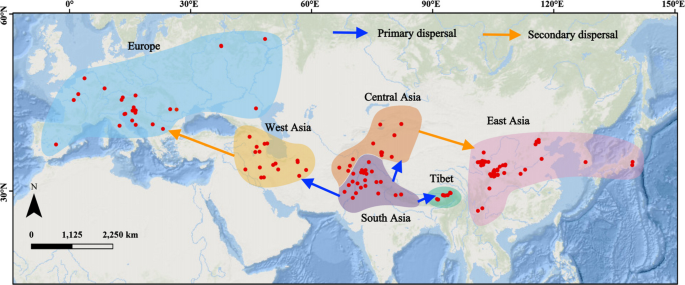
核心成果提炼:该Biomarker明确了南亚(喜马拉雅西部及周边区域)作为普通核桃驯化起源地的地位,创新性地将方法学偏倚校正引入作物起源研究,解决了长达数十年的核桃起源争议。统计学结果显示,南亚与其余群体的遗传分化系数为0.074-0.289,其中与西亚的遗传分化系数最低(0.074,n=16/51,P<0.001),支持从南亚向西亚的扩散路径。基于该Biomarker进一步明确了核桃的全球扩散路线:从南亚分别向东经中亚扩散到东亚,向南经高原南路扩散到西藏,向西经西亚扩散到欧洲(对应文献图6)。