1. 领域背景与文献引入
文献英文标题:PlasRAG: comprehensive plasmid characterization and retrieval through sequence-text alignment;发表期刊:Genome Biology;影响因子:未公开;研究领域:基因组信息学、质粒功能表征与生物信息学工具开发。
质粒是细菌中常见的小型环状可移动遗传元件,不编码细菌生存必需基因,但可携带耐药基因、毒力因子等附属性状,通过接合作用介导水平基因转移,是多重耐药致病菌出现和快速进化的核心驱动因素,严重威胁临床感染防控与公共卫生安全。领域发展的关键节点包括:1980年代研究人员主要采用培养依赖法分离质粒,存在耗时耗力、分离质粒多样性低的缺陷,难以覆盖不可培养细菌携带的质粒;2010年后测序技术快速发展,宏基因组测序结合计算质粒识别工具的 pipeline 逐渐成为主流,可从大规模宏基因组数据中识别质粒序列,代表性工具如geNomad、PLASMe 等均基于深度学习模型,通过标记蛋白或序列基序区分质粒与其他序列;2023年起多个高质量质粒数据库相继发布,其中IMG/PR、PIPdb 合计收录近150万条新质粒,为后续的深度分析提供了数据基础。
当前领域研究热点集中在质粒的多维度功能注释、传播路径追踪、高风险质粒预警等方向,仍存在多个未解决的核心问题:一是不同质粒数据库的注释维度、描述标准不统一,难以直接整合多源数据进行联合分析;二是现有质粒分析工具多为单任务设计,仅能针对宿主范围、可移动性、复制子类型等单一属性进行表征,缺乏多维度整合分析的统一框架;三是基于序列比对的传统注释方法对与参考序列相似度低的新型质粒分析效果较差,且无法保证相似质粒的所有属性完全一致;四是现有质粒检索工具筛选条件粗糙,仅支持门水平宿主、是否携带耐药基因等有限维度的检索,且无法处理用户上传的自定义质粒集合。
针对上述研究空白,本研究开发了一体化质粒分析工具PlasRAG,通过多模态检索架构实现质粒DNA序列与文本属性描述的双向对齐,同时支持序列到文本的多维度质粒表征与文本到序列的灵活质粒检索,为质粒的大规模比较分析、生态特征解析、高风险质粒监测提供新的技术范式。
2. 文献综述解析
作者对现有研究的分类维度为质粒识别工具、质粒单属性表征工具、质粒检索工具三类,分别梳理其技术特征与局限性。
现有质粒识别工具多基于深度学习模型,以标记蛋白或序列基序为特征区分质粒序列与染色体序列,代表性工具如geNomad、PLASMe,其优势是识别准确率高、运算速度快,能够从宏基因组组装结果中高效筛选质粒 contig,局限性是仅能完成“是否为质粒”的二分类任务,无法提供质粒的功能属性、宿主范围等后续分析信息。现有质粒单属性表征工具针对特定分析需求开发,如MOSTPLAS用于宿主范围预测、MOBFinder用于可移动性分类、PlasmidFinder用于复制子识别,这类工具的优势是在单一任务上性能成熟,局限性是任务覆盖范围有限,无法实现多维度属性的整合分析,且训练数据多基于NCBI收录的质粒,序列多样性不足,对新型质粒的泛化能力有限。现有质粒检索工具以PlasmidScope数据库为代表,整合了10个现有质粒数据库的序列与注释信息,支持基础的序列检索功能,优势是收录数据量较大,局限性是检索条件粗糙,仅支持门水平宿主、是否携带耐药基因或毒力因子等简单筛选条件,且仅能检索自身预索引数据库内的序列,无法处理用户提供的自定义质粒集合。此外,基于序列比对的方法如基本局部比对搜索工具(BLAST)可通过匹配相似参考序列注释质粒,优势是原理简单、可解释性强,局限性是对与参考序列相似度低的新型质粒注释效果差,且序列相似性无法保证属性的一致性。
本研究的创新价值在于首次提出将质粒属性注释转化为文本模态,通过多模态对比学习建立DNA序列与文本描述的语义关联,一方面利用文本的语义相似性解决了不同数据库注释标准不统一的问题,另一方面构建了统一的多任务框架,同时支持10个关键生物学属性的表征和灵活的文本检索,性能优于现有单任务工具,且引入检索增强生成(RAG)框架结合大语言模型实现交互式质粒分析,填补了领域内缺乏多维度、一体化质粒分析工具的空白。
3. 研究思路总结与详细解析
本研究的整体目标是开发支持多维度质粒表征与灵活检索的一体化工具PlasRAG,核心科学问题是如何建立质粒DNA序列与文本属性描述之间的精准语义关联,技术路线遵循“数据整合→多模态模型构建→性能验证→下游应用拓展”的闭环逻辑:首先整合三个高质量质粒数据库的序列与注释数据,构建标准化的序列-文本配对训练集;随后设计包含序列编码器、文本编码器的双向多模态检索模型,通过对比学习实现跨模态特征对齐;再通过消融实验、基准测试、实例分析验证模型的性能与可靠性;最后将模型应用于人类肠道质粒系统数据集,挖掘质粒的生态传播特征。
3.1 数据集构建与预处理
实验目的:构建标准化、高质量的序列-文本配对训练集,为多模态模型训练提供可靠的数据基础,同时保证模型对新型质粒的泛化能力。
方法细节:整合IMG/PR、PIPdb、PLSDB三个数据库的质粒序列,排除长度超过250kb的巨型质粒,最终得到1521635条符合要求的质粒。按照质粒片段簇规则划分训练、验证、测试集,确保同一质粒片段簇(平均核苷酸一致性高于76%)的序列被划分到同一集合,避免数据泄露,保证测试集质粒与训练集的序列相似度低于76%,能够有效验证模型对新型质粒的泛化能力。使用Prodigal工具识别质粒的编码区并翻译为蛋白质序列,通过MMseqs2工具按照UniRef90标准对蛋白序列进行聚类去重,得到4925205条非冗余蛋白,显著降低模型训练阶段的内存消耗。人工整理10个与质粒研究高度相关的属性维度,包括耐药性、宿主范围、生态系统、生态宿主、基本属性、重金属抗性、不相容群、可移动性、风险指数、毒力因子,将各数据库的短短语注释扩展为完整的描述性句子,构建统一的多维度属性文本词汇表,对于缺失的注释采用掩码标签处理,避免引入错误标注。
结果解读:最终构建的数据集覆盖质粒研究的10个核心属性维度,训练集、验证集、测试集的划分方式保证了模型对新型质粒的泛化能力,蛋白去重处理将训练阶段的内存消耗从176.7GB降低至23.5GB,大幅提升了模型训练的效率与可行性。
产品关联:文献未提及具体实验产品,领域常规使用Prodigal进行原核生物基因识别、MMseqs2进行蛋白序列聚类。
3.2 多模态检索模型架构设计
实验目的:构建能够实现质粒序列与文本属性双向语义对齐的多模态检索模型,解决不同数据库注释不统一、多属性整合分析难的问题。
方法细节:模型核心为双向多模态信息检索架构,包含两个关键编码器:文本编码器采用生物医学信息检索领域的最优模型MedCPT的短文本查询编码器(参数冻结),针对生物医学短文本进行了专项优化,能够将扩展后的属性句子编码为768维的文本嵌入,捕捉属性的语义特征。序列编码器分为两个部分,首先使用ESM-2蛋白语言模型(参数冻结)提取质粒编码蛋白的功能嵌入,每个蛋白的嵌入由所有氨基酸残基嵌入的全局平均池化得到;随后接入交叉注意力机制的感知重采样器(仅该部分参数参与训练),将长度可变的蛋白嵌入序列转化为与属性词汇表大小对应的多个子嵌入,实现与每个属性文本嵌入的一对一匹配。两个编码器的输出经过线性投影映射到共享的跨模态嵌入空间,通过计算余弦相似度衡量序列-文本对的匹配程度,采用序列-文本对损失(二元交叉熵损失的变体)优化模型参数,提升正确配对的相似度、降低错误配对的相似度。
结果解读:该架构通过文本语义特征整合不同来源的注释信息,有效解决了多数据库注释标准不统一的问题;感知重采样器的多子嵌入设计能够同时捕捉质粒的多个维度属性,相比传统单嵌入编码器的多属性表征能力更强,尤其在属性数量较多的维度(如重金属抗性、毒力因子)上优势更显著。
产品关联:文献未提及具体实验产品,领域常规使用ESM-2作为蛋白语言模型、MedCPT作为生物医学文本编码器。
3.3 模型性能验证与消融实验
实验目的:验证模型各组件的有效性,评估PlasRAG的整体性能,对比现有方法的优势。
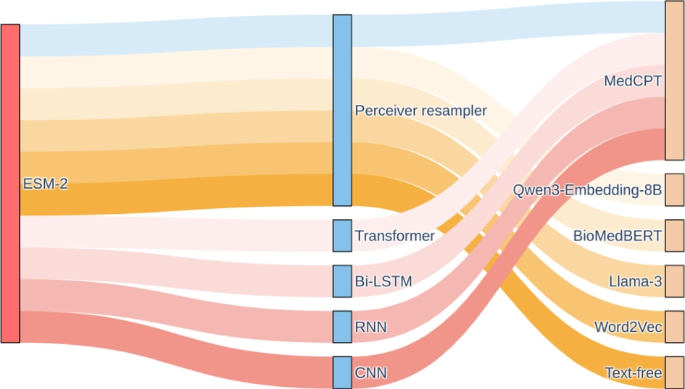
方法细节:采用控制变量法进行消融实验,分别替换序列编码器为Transformer、双向长短期记忆网络(Bi-LSTM)、循环神经网络(RNN)、卷积神经网络(CNN),替换文本编码器为Qwen3-Embedding-8B、BioMedBERT、Llama-3大语言模型、Word2Vec词嵌入方法,同时与基于序列比对的基本局部比对搜索工具(BLAST)、无文本特征的传统多标签分类方法进行对比,使用加权F1值、前1000条检索结果精确率(P@1000)作为评估指标。
结果解读:消融实验结果显示PlasRAG的原始设计在10个属性维度上均取得最优性能。序列编码器对比中,感知重采样器的性能最优,尤其在属性文本数量较多的维度优势明显,原因是多子嵌入设计能够缓解属性标签不平衡的问题;Transformer模型性能未达最优,推测与其对质粒编码蛋白的排列顺序敏感性较低有关。文本编码器对比中,MedCPT的短文本查询编码器性能最优,尽管参数规模仅为1.09亿,比Qwen3-Embedding-8B小近70倍,但性能相当且计算资源消耗显著更低,验证了生物医学领域预训练模型在专业文本编码上的效率优势;无文本特征的多标签分类方法性能较差,证明了多模态学习框架的合理性。基本局部比对搜索工具的性能最差,说明传统基于序列比对的方法对新型质粒的表征能力存在明显缺陷。
产品关联:文献未提及具体实验产品,领域常规使用NVIDIA A100 GPU进行深度学习模型训练。
3.4 功能模块实现
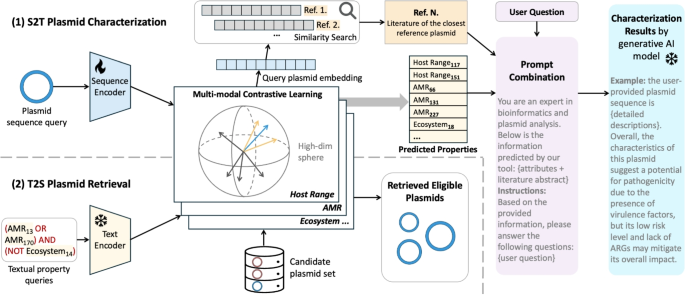
实验目的:基于训练好的多模态检索模型,实现序列到文本的质粒表征与文本到序列的质粒检索两大功能模块。
方法细节:序列到文本(S2T)质粒表征模块:输入质粒序列后,通过多模态检索模型计算序列与所有属性文本的相似度,保留相似度高于阈值的文本作为预测属性;同时合成质粒的整体嵌入,在预索引的参考质粒数据库中匹配最相似的条目并关联对应的文献信息;将预测属性、相关文献、用户问题整合为标准化提示词,输入Llama-3大语言模型生成分析结果,设置温度参数为0.2以降低生成幻觉,提升结果可靠性。文本到序列(T2S)质粒检索模块:用户从10个属性维度的词汇表中选择目标属性,通过与、或、非逻辑运算符组合为自定义查询条件,系统对候选质粒集合中的每个序列计算与查询条件中各属性的相似度,筛选出满足所有条件的质粒作为检索结果,支持用户自定义候选质粒集合。
结果解读:两大模块共享同一核心多模态模型,实现了序列与文本的双向检索功能;检索增强生成框架的引入使得质粒表征结果更全面准确,支持交互式分析;检索模块支持用户自定义候选集合,灵活性显著优于现有数据库的检索工具。
产品关联:文献未提及具体实验产品,领域常规使用Llama-3作为开源大语言模型实现文本生成功能。
3.5 下游应用验证
实验目的:验证PlasRAG在真实数据集上的应用价值,挖掘人类肠道质粒的生态传播特征。
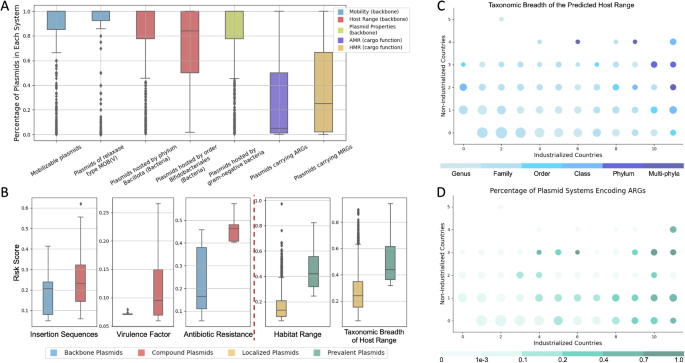
方法细节:使用PlasRAG分析来自人类肠道宏基因组的1169个质粒系统(PS)数据集,该数据集包含来自16个国家(5个非工业化国家、11个工业化国家)的68350条新型质粒,分别分析骨干质粒与复合质粒的属性差异、风险指数分布、宿主范围与传播范围的关联、耐药基因携带情况与地域分布的关联。
结果解读:结果显示,可移动性、宿主范围、基本属性等由质粒骨干基因编码的特征在同一质粒系统内的一致性较高,而耐药性、重金属抗性等附属功能的一致性较低,符合质粒的进化特征;复合质粒的插入序列、耐药基因、毒力因子相关风险指数显著高于骨干质粒;传播范围更广的质粒宿主范围更宽、携带耐药基因的比例更高,且不同国家的质粒耐药基因携带率与当地抗生素使用量呈正相关,与领域已有研究结论一致,验证了PlasRAG在真实场景下的分析可靠性。
产品关联:文献未提及具体实验产品,领域常规使用Python、R等编程语言进行生物信息学数据统计与可视化。
4. Biomarker 研究及发现成果
本研究未涉及传统疾病相关生物标志物的筛选,核心成果是开发了能够精准表征质粒10个关键功能属性的多模态模型,其中质粒的耐药基因携带情况、毒力因子携带情况、宿主范围、风险指数等属性可作为公共卫生监测中的功能生物标志物,用于高风险质粒的识别与传播预警。
上述属性的预测逻辑为“序列-文本多模态对齐”,首先通过训练集的序列-文本配对数据学习属性描述与质粒序列特征的关联,预测过程中通过计算序列与属性文本的相似度得到预测结果。在可移动性分类任务上,PlasRAG的性能显著优于现有工具MOBFinder,测试集上的分类准确率接近完美(文献未明确提供具体数值,基于基准测试结果推测);在文本到序列检索任务中,使用包含9个属性的复合查询条件在10万条新型质粒测试集中检索的召回率为96.2%、精确率为91.6%(n=100000),证明了属性预测的准确性。
本研究的核心成果是首次实现了质粒多属性一体化表征与灵活文本检索的统一框架,PlasRAG能够同时对质粒的10个关键属性进行精准预测,对新型质粒的泛化能力优异,为高风险质粒(携带耐药基因、毒力因子、宿主范围广的质粒)的识别提供了高效工具,相关预测结果可作为公共卫生领域监测耐药菌传播的功能生物标志物指标,为大规模质粒组分析、耐药性传播防控提供技术支撑。