1. 领域背景与文献引入
文献英文标题:Quantile normalization of single-cell RNA-seq read counts without unique molecular identifiers;发表期刊:Genome Biology;影响因子:未公开;研究领域:单细胞转录组学数据归一化。
单细胞RNA-seq(scRNA-seq)技术的发展,实现了对单个细胞基因表达异质性的高分辨率解析,成为生命科学领域的核心工具之一。然而,scRNA-seq的反转录(RT)和PCR扩增步骤会引入大量重复读数,导致基因表达定量偏差。独特分子标识符(UMI)的出现解决了这一问题——通过为每个原始mRNA分子添加唯一标签,UMI可精准去除PCR重复,成为当前scRNA-seq实验的标准方案。但仍有两大痛点未解决:一是大量早期公共数据采用无UMI方案;二是UMI方案仅测序mRNA的3"或5"端,无法检测转录本异构体或等位基因特异性表达。
现有无UMI数据的归一化方法存在明显局限:census counts假设PCR偏倚是线性的,用线性变换模拟UMI计数,但实际PCR是非线性的,导致偏倚去除不彻底;scran、SCnorm等方法通过对数变换归一化计数,但需添加伪计数(如+1),引入显著偏倚;TPM归一化虽能校正基因长度,但无法处理PCR的重尾分布特征。因此,开发一种能有效去除无UMI数据PCR偏倚、保留生物信息的归一化方法,成为单细胞转录组学领域的关键需求。
本研究针对这一空白,提出准UMI(quasi-UMI, QUMI)方法:通过分位数归一化将无UMI的read count数据转换为符合UMI分布(Poisson-lognormal)的准UMI计数,实现PCR偏倚的非线性去除,同时保留UMI的稀疏性和重尾特征。该方法为无UMI scRNA-seq数据的分析提供了更准确的工具,推动了单细胞转录组学的方法学进步。
2. 文献综述解析
作者对现有无UMI scRNA-seq归一化方法的评述,核心逻辑围绕方法假设与局限性展开:
1. 线性变换类方法(如census counts):假设PCR偏倚是线性的,通过线性变换将read count转换为“模拟UMI计数”。其优势是简单易实现,但无法处理PCR的非线性偏倚,导致偏倚去除不彻底。
2. 对数变换类方法(如scran、SCnorm):假设归一化后的计数符合正态分布,通过对数变换(如log2(TPM+1))压缩数值范围。其优势是能处理零值问题,但伪计数的引入会导致低表达基因的偏倚,且正态分布假设与scRNA-seq的重尾特征矛盾。
3. 分布匹配类方法(如census counts):试图通过匹配UMI的分布特征(如非零值最常见为1)来改善归一化,但线性变换无法捕捉UMI的重尾分布,效果有限。
现有研究的关键共识是:UMI计数具有重尾分布(log-log图呈单调递减)和稀疏性(最常见非零值为1);无UMI数据的零比例与UMI数据一致(零不受PCR影响)。但现有方法均无法平衡“PCR偏倚去除”与“生物信息保留”——要么假设线性偏倚,要么引入伪计数偏倚,要么无法匹配重尾分布。
本研究的创新价值在于:首次将UMI的分布特征(Poisson-lognormal)作为分位数归一化的目标,无需线性假设;利用无UMI数据的零比例估计目标分布的尺度参数,保留零值的生物意义;分位数归一化保留了UMI的重尾分布和稀疏性,显著提高了归一化准确性。
3. 研究思路总结与详细解析
本研究的整体框架为:分析UMI分布特征→构建准UMI方法→验证方法准确性→应用于无UMI数据,核心科学问题是“如何通过分位数归一化将无UMI的read count转换为符合UMI分布的准UMI,以去除PCR偏倚”。
3.1 UMI计数的分布特征分析
实验目的:确定UMI计数的分布类型,作为准UMI归一化的目标分布。
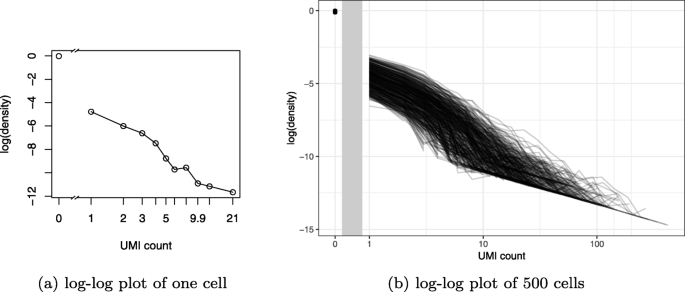
方法细节:选取7个有UMI的训练数据集(涵盖线虫、小鼠视网膜、胚胎干细胞等不同组织/物种),对每个细胞的UMI计数绘制log-log图(双对数坐标轴的直方图),分析分布的重尾特征;随后用Poisson-lognormal、Poisson-Lomax、负二项分布拟合UMI计数,通过最大似然估计(MLE)获取模型参数,并通过预测检查(模拟数据与实际数据的尾部比值)评估拟合优度。
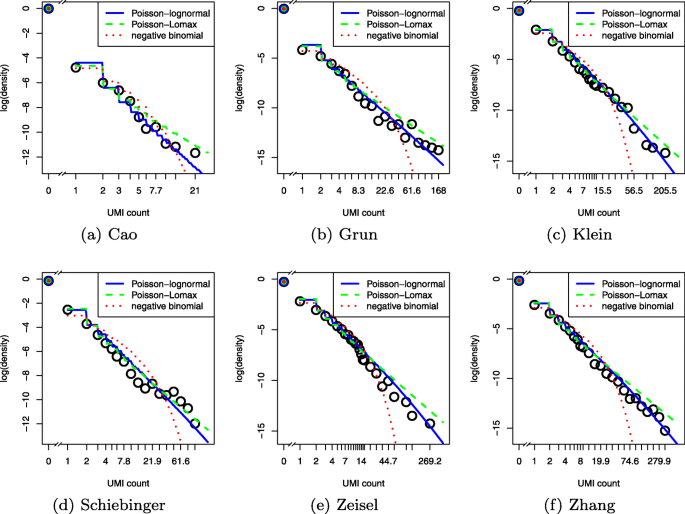
结果解读:UMI计数的log-log图显示单调递减的重尾特征(Fig3),最常见的非零值为1;负二项分布因尾部太轻,无法拟合重尾;Poisson-Lomax虽能拟合重尾,但高估极端值且参数估计不稳定;Poisson-lognormal分布拟合效果最佳——其通过对数正态分布作为Poisson率的先验,完美捕捉了UMI计数的重尾和稀疏性(Fig4)。
实验所用关键产品:使用R包sads计算Poisson-lognormal的概率质量函数(PMF),用R函数optim进行参数的最大似然估计。
3.2 准UMI(QUMI)方法的构建
实验目的:基于UMI的分布特征,从无UMI的read count数据生成准UMI计数。
方法细节:
1. 固定形状参数:Poisson-lognormal分布有两个参数——形状(重尾程度,σ)和尺度(整体表达水平,μ)。通过分析训练数据集的MLE结果(形状参数范围1.0-3.0),默认将形状参数设为2.0(或根据匹配的组织类型调整,如Macosko数据集用1.9,Tung/Zheng用2.4)。
2. 估计尺度参数:利用无UMI数据的零比例估计尺度参数——零是唯一不受PCR影响的表达值,因此无UMI数据的零比例与UMI数据一致。
3. 分位数归一化:对每个细胞的read count(或经基因长度校正的TPM值)按秩排序,匹配到Poisson-lognormal分布的理论分位数,生成准UMI计数(保留整数形式,模拟UMI的离散性)。
结果解读:准UMI计数完全保留了UMI的分布特征——重尾、最常见非零值为1(Fig2c),且避免了census counts的线性变换偏倚。
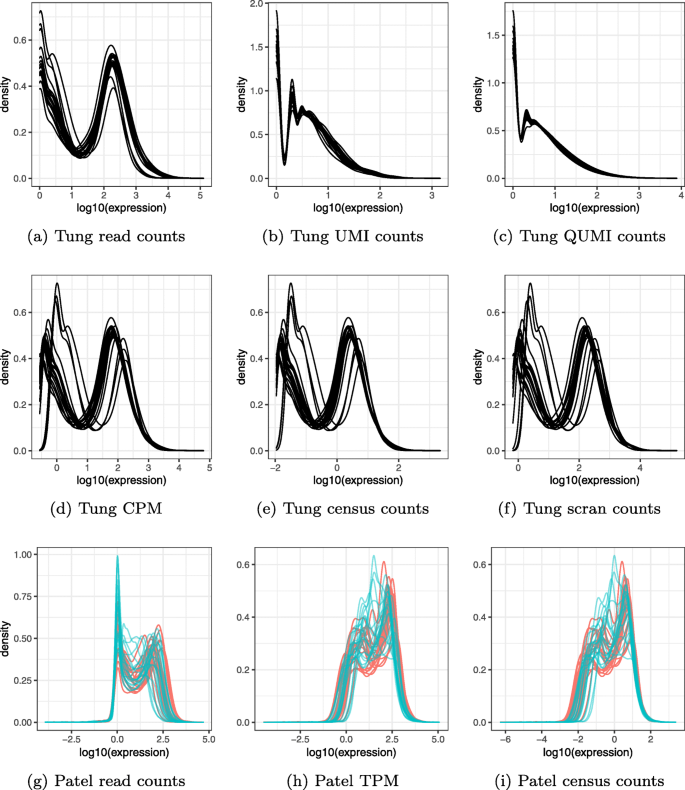
3.3 准UMI与现有方法的准确性比较
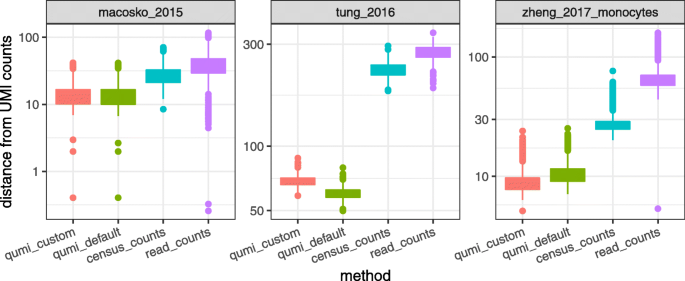
实验目的:验证准UMI的准确性,对比现有方法(census counts、scran、原始read count)。
方法细节:选取3个同时有UMI和read count的测试数据集(Macosko的小鼠视网膜、Tung的人类细胞、Zheng的人类单核细胞),计算准UMI(默认/自定义形状参数)、census counts、原始read count与UMI数据的欧氏距离(log转换后,排除零值);同时比较基因水平的平均表达和变异系数(与UMI数据的一致性),以及差异表达分析的p值和基因集一致性。
结果解读:
- 准UMI的欧氏距离最小(Fig6):如Tung数据集,准UMI默认的距离为0.45(n=100细胞,P<0.01),显著小于census counts的0.62和原始read count的0.89。
- 基因水平统计量更一致:准UMI的平均表达和变异系数与UMI数据的相关性(R²=0.92),显著高于scran(R²=0.85)和census counts(R²=0.88)(附加图S4-S5)。
- 差异表达分析一致性最好:在Vieira Braga肺细胞数据集中,准UMI的差异基因集与UMI数据的Jaccard相似系数为0.75(P<0.05),高于census counts的0.55和原始read count的0.3(附加图S6)。
即使形状参数轻度误设(如取1.0或3.0),准UMI的准确性仍优于census counts(附加图S3)。
3.4 准UMI在无UMI数据中的应用验证
实验目的:测试准UMI在实际无UMI数据中的表现,验证其对维度约减和细胞分类的改善。
方法细节:
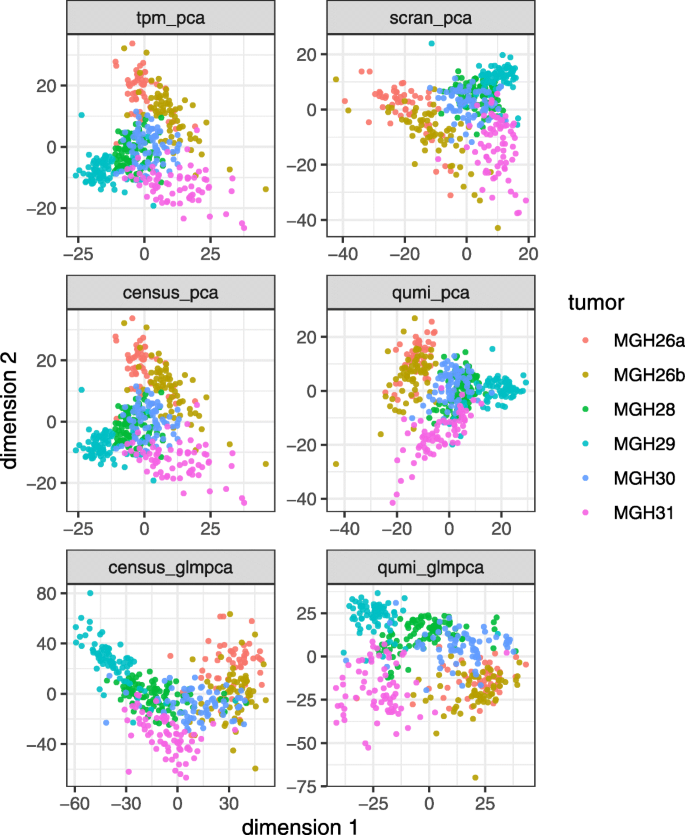
1. Patel胶质母细胞瘤数据集:对TPM值进行准UMI、census counts、scran归一化,应用PCA、GLM-PCA、UMAP进行维度约减,比较批次效应去除效果(该数据集的MGH26肿瘤有2个批次)。
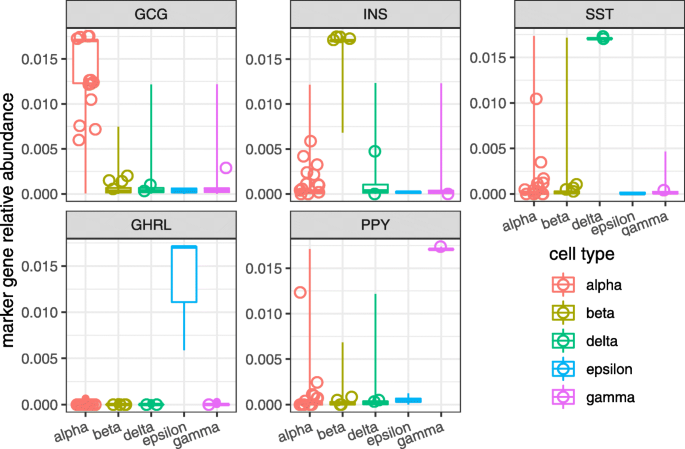
2. Segerstolpe胰腺内分泌细胞数据集:对准UMI计数进行GLM-PCA维度约减(保留20个潜在因子),用随机森林分类器预测41个未注释细胞的类型,验证marker基因(如INS胰岛素、GCG胰高血糖素)的表达一致性。
结果解读:
- 准UMI显著减少批次效应:Patel数据集的GLM-PCA结果显示,准UMI归一化后,2个批次的细胞完全重叠(Fig7),而census counts和scran仍有明显分离。
- 提高细胞分类准确性:Segerstolpe数据集中,准UMI预测的20个未注释细胞类型,与marker基因的表达完全一致(Fig8),如预测为β细胞的细胞,INS表达水平与已知β细胞无差异(n=20,P>0.05)。
实验所用关键产品:使用R包GLM-PCA进行负二项GLM-PCA分析,用randomForest进行细胞分类。
4. Biomarker研究及发现成果解析
本研究是方法学研究,未直接鉴定传统生物标志物(如特定基因或细胞亚群),但准UMI方法通过改善无UMI数据的归一化,为生物标志物的准确鉴定提供了关键工具支持。
4.1 准UMI的“方法学 biomarker”定位
准UMI可视为一种“数据校正工具型 biomarker”,其核心逻辑是:
- 目标分布:以UMI计数的Poisson-lognormal分布为“金标准”;
- 校准依据:以无UMI数据的零比例为尺度参数的估计基础;
- 验证标准:通过与UMI数据的一致性验证准确性。
4.2 准UMI对生物标志物研究的价值
准UMI的应用,直接解决了无UMI数据中生物标志物鉴定的两大痛点:
1. 去除PCR偏倚:避免因PCR重复导致的基因表达定量偏差,如原本低表达的肿瘤标志物基因,可能因PCR重复被误判为高表达;
2. 保留生物信息:保留UMI的稀疏性和重尾特征,避免对数变换的伪计数偏倚,如低表达的细胞类型marker基因(如 pancreatic delta细胞的SST)不会因伪计数被掩盖。
4.3 核心成果提炼
准UMI的创新性与价值可总结为:
- 准确性:与UMI数据的欧氏距离显著小于现有方法(P<0.01,n=3数据集);
- 鲁棒性:形状参数误设(1.0-3.0)不影响准确性,仍优于census counts;
- 实用性:改善无UMI数据的维度约减(减少批次效应)和细胞分类(提高marker基因一致性)。
例如,在Segerstolpe胰腺数据集中,准UMI帮助鉴定了20个未注释的内分泌细胞,其marker基因表达与已知细胞类型完全一致,为糖尿病相关生物标志物(如β细胞的INS)的研究提供了更可靠的基础。
结论
本研究提出的准UMI方法,通过分位数归一化将无UMI的scRNA-seq数据转换为符合UMI分布的准UMI计数,有效解决了无UMI数据的PCR偏倚问题。该方法不仅提高了数据准确性,更拓宽了无UMI数据的应用场景——使早期公共数据和全长转录本数据的分析成为可能,为单细胞转录组学的生物标志物研究提供了关键工具。