1. 领域背景与文献引入
文献英文标题:The first Irish genome and ways of improving sequence accuracy;发表期刊:Genome Biology;影响因子:未公开;研究领域:人类基因组学、个性化医疗。
领域共识:2000年人类基因组草图的发布开启了人类基因组学研究的新纪元,随后dbSNP数据库持续积累人类遗传变异数据,2009年版本已包含约1390万个单核苷酸多态性(SNP)和450万个插入缺失变异(indel)。下一代测序技术的突破性进展大幅降低了测序成本与时间,推动个人基因组测序成为研究热点,截至2010年已有至少15个非癌症个人基因组通过不同平台完成测序。当前领域的核心研究方向是将遗传变异信息转化为个性化医疗的基础,但仍面临诸多未解决问题,包括人类基因组中未知变异的类型与数量、高效检测所有变异所需的群体规模与个体数量、不同频率变异在个体基因组中的分布,以及直接影响疾病风险的功能变异鉴定等。
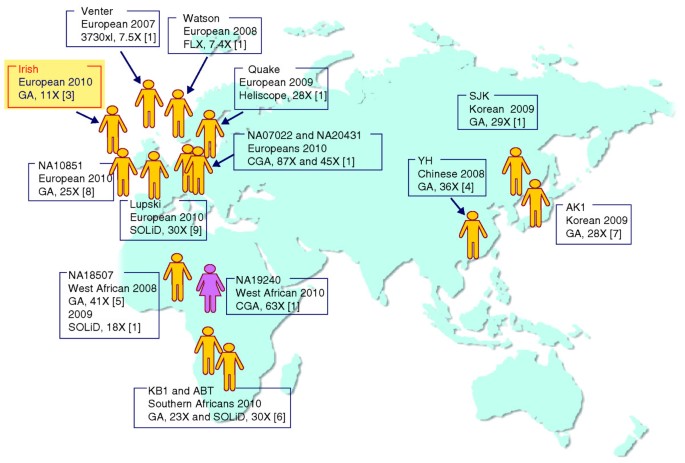
爱尔兰人群因地处欧洲西部边缘且长期地理隔离,被认为可能携带大量人群特异性遗传多态性,是基因组研究的理想对象。本文作为首个爱尔兰人全基因组测序研究,不仅填补了欧洲隔离人群的基因组数据空白,还针对低深度测序的准确性瓶颈提出了基于基因型填充的解决方案,为平衡测序资源与数据准确性提供了新的技术范式,对推动大规模人群基因组研究具有重要学术价值。
2. 文献综述解析
本文综述部分以测序技术平台、人群地域分布、测序覆盖度为分类维度,系统梳理了2010年之前的个人基因组测序研究,明确了现有研究的优势、局限性与未解决问题。
现有研究的关键结论是全基因组测序是检测新型遗传变异的最有效手段,不同测序平台与覆盖度的组合会显著影响变异检测的结果;技术方法层面,下一代测序技术的优势在于低成本与高速度,可在短时间内完成个人基因组测序,但低覆盖度(如11X)测序存在明显局限性,包括杂合变异的漏检(假阴性)与误判(假阳性),导致变异检测的敏感性与阳性预测值下降,而高覆盖度(20X以上)测序虽能提升准确性,但需要投入更多资源。现有研究多集中在非隔离人群,对欧洲边缘隔离人群的基因组变异特征了解不足,同时缺乏在有限资源下提升低深度测序准确性的有效方法。
通过对比现有研究的局限性,本文的创新点凸显为两个方面,一是首次完成爱尔兰隔离人群的全基因组测序,鉴定出大量人群特异性的新型遗传变异,补充了人类基因组变异图谱的群体特异性数据;二是首次系统验证了利用已有群体基因型数据进行填充的方法,可在低深度测序(5X甚至2X)下获得95%以上的基因型准确性,解决了大规模人群基因组研究中资源与准确性的平衡问题,为后续研究提供了可借鉴的技术路线。
3. 研究思路总结与详细解析
本文的研究目标是完成首个爱尔兰人全基因组测序并分析其遗传变异特征,同时探索低深度测序准确性的提升策略;核心科学问题包括隔离人群的基因组变异特征,以及如何在有限资源下保障低深度测序的数据质量;技术路线遵循“样本测序→变异分析→局限性评估→方法优化→进化分析”的逻辑闭环,从基础数据产出到应用方法探索形成完整的研究链条。
3.1 爱尔兰人全基因组测序与变异检测
实验目的是获取首个爱尔兰人全基因组的完整序列信息,系统鉴定其中的遗传变异类型与数量。方法细节为采用Illumina Genome Analyzer测序平台,对爱尔兰人样本进行全基因组测序,生成4.4亿条短读长序列,最终获得全基因组11X的测序覆盖度。结果解读显示,本次测序共检测到超过300万个SNP,其中约13%(40万个)为dbSNP数据库130版本中未收录的新型变异,同时鉴定出约20万个短indel,其中一半为首次报道的新型变异;在巨噬细胞刺激1(MST1)基因中发现一个新的非同义SNP,推测该变异可能通过影响蛋白功能参与炎症性肠病的发病机制。
文献未提及具体实验产品,领域常规使用Illumina系列测序试剂盒、基因组变异检测软件(如基因组分析工具包GATK)等。
3.2 低深度测序局限性评估
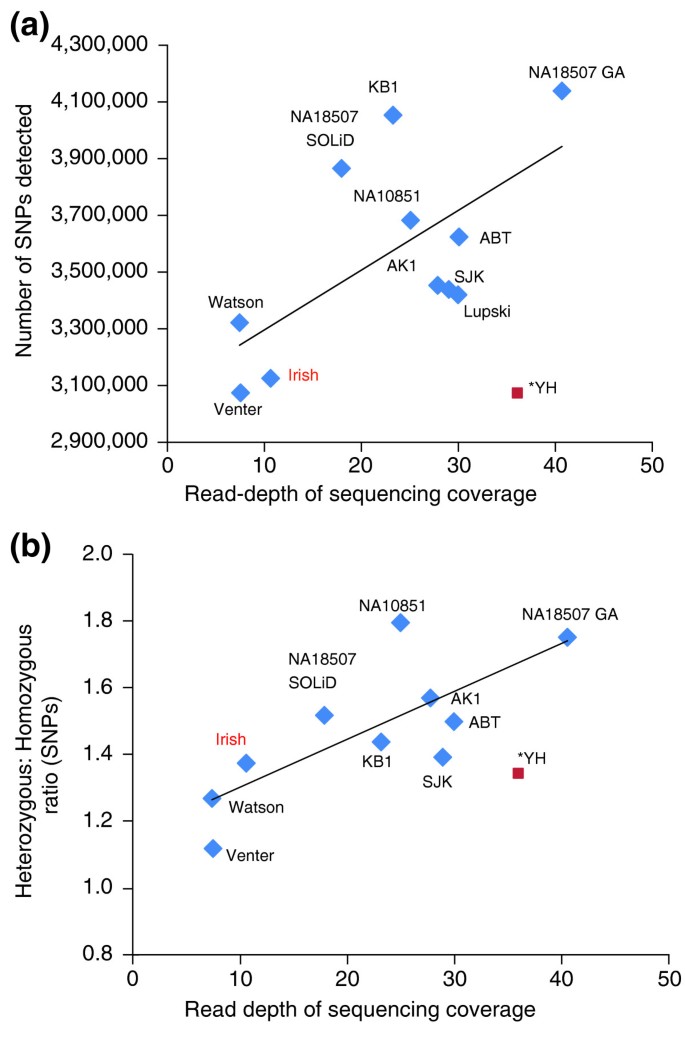
实验目的是明确低覆盖度测序对遗传变异检测准确性与完整性的影响。方法细节为将本次11X覆盖度的爱尔兰基因组数据,与已发表的20X以上覆盖度的欧洲人群个人基因组(如NA10851、Lupski)进行对比,分析SNP检测数量与假发现率的差异。结果解读显示,爱尔兰基因组的SNP检测数量比高深度测序的样本少约30万个,假发现率为1.4%,而高深度测序可将假发现率降至0.1%以下;进一步分析表明,低深度测序更易漏检杂合变异,随着测序覆盖度的提升,杂合变异的检测比例显著增加。
文献未提及具体实验产品,领域常规使用变异数据集对比工具(如VCFtools)、统计分析软件等。
3.3 基因型填充提升低深度测序准确性验证
实验目的是验证利用已有群体基因型数据填充,提升低深度测序基因型准确性的可行性与效果。方法细节为整合HapMap项目与1000 Genomes项目的欧洲人群基因型数据,分别对5X与2X覆盖度的模拟低深度测序数据进行基因型填充分析,评估填充后的基因型准确性。结果解读显示,5X覆盖度测序结合基因型填充可达到99%以上的基因型准确性,即使是2X覆盖度测序,结合填充后也能获得95%以上的准确性,证明该方法可在大幅降低测序资源投入的同时,保障数据的可靠性。文献未提及具体实验产品,领域常规使用基因型填充软件(如IMPUTE2、BEAGLE)等。
3.4 全基因组正选择信号分析
实验目的是探索爱尔兰人基因组中受到自然选择的基因区域,揭示隔离人群的进化特征。方法细节为结合本次测序数据与已发表的9个个人基因组数据,采用群体遗传学分析方法,检测基因组中的正选择与平衡选择信号。结果解读显示,在嗅觉与味觉受体基因位点检测到平衡选择信号,与之前的全基因组SNP研究结果一致,提示这些基因的多态性可能与人群的环境适应相关;同时在近期发生基因复制的区域检测到正选择信号,为人类进化过程中的基因功能创新提供了新的证据。文献未提及具体实验产品,领域常规使用群体遗传学分析工具(如PLINK、Fst计算软件)等。
4. Biomarker研究及发现成果
本文涉及的Biomarker为爱尔兰人群特异性的遗传变异,包括新型SNP与indel,其筛选逻辑为通过全基因组测序鉴定dbSNP数据库未收录的变异,结合爱尔兰人群的地理隔离特征,判断这些变异为该人群所特有,验证过程遵循“全基因组检测→数据库对比→人群特异性分析”的完整链条。
这些Biomarker的样本来源为爱尔兰人的外周血基因组DNA,验证方法为全基因组测序与dbSNP数据库的比对分析,同时通过与其他欧洲人群的基因组数据对比,确认其人群特异性;特异性方面,约13%的SNP与50%的indel为爱尔兰人群首次报道的新型变异(文献未明确提供敏感性数据)。
本次研究共发现40万个新型SNP与10万个新型indel,其中MST1基因的非同义SNP具有潜在的疾病关联价值,推测可作为炎症性肠病的人群特异性风险评估Biomarker;研究的创新性在于首次系统鉴定了欧洲隔离人群的特异性遗传变异,为复杂疾病的群体遗传学研究提供了新的候选Biomarker;文献未明确提供该Biomarker的统计学显著性数据(如P值、风险比等),其功能与疾病关联仍需后续实验验证。