1. 领域背景与文献引入
文献英文标题:FRESCo: finding regions of excess synonymous constraint in diverse viruses;发表期刊:Genome Biology;影响因子:未公开;研究领域:病毒基因组功能元件注释、进化生物学
病毒基因组因空间极度紧凑,需在有限长度内编码完成入侵、复制、包装、释放的全部信息,因此重叠功能元件是其常见特征,包括微小RNA、重叠阅读框、转录调控位点、包装信号等。传统观点认为蛋白编码区的同义突变是中性的,但近年研究证实同义突变常具有重要功能意义,可影响mRNA二级结构、转录效率、翻译准确性等。随着测序技术发展,部分病毒的分离株序列数量已达数千株,为检测病毒基因组中的进化约束区域提供了数据基础,但现有检测方法存在分辨率低、缺乏通用实现工具、统计模型不严谨等问题,无法高效利用深度序列比对数据解析短病毒基因组中的重叠功能元件。本研究针对这一领域空白,开发了基于系统发育密码子模型的FRESCo方法,实现了高分辨率检测病毒基因中的同义约束过剩区域,为病毒基因组功能注释提供了新的技术工具。
2. 文献综述解析
作者按研究对象与方法局限性对现有研究进行分类梳理,核心评述逻辑围绕“同义突变的功能意义→病毒重叠元件的研究进展→现有检测方法的不足”展开。现有研究已在细菌、昆虫、哺乳动物及病毒中证实同义突变的功能价值,病毒基因组中已发现多种类型的重叠功能元件,但相关研究多聚焦于特定病毒,缺乏可推广的通用检测工具;已有的同义约束检测方法如Mayrose等人的模型仅应用于HIV基因组,且无公开可用的实现方案,其他方法多为低分辨率检测,缺乏严谨的统计模型比较框架,无法实现单密码子水平的高分辨率分析。本研究的创新价值在于,首次将适用于哺乳动物基因组的系统发育密码子模型适配到病毒短基因组的深度比对数据中,开发了具备统计检验支撑的FRESCo方法,不仅能实现单密码子分辨率的约束区域检测,还提供了公开可用的HYPHY批处理脚本,可应用于任意开放阅读框的比对数据,弥补了现有方法的局限性。
3. 研究思路总结与详细解析
本研究的核心目标是开发并验证一种高分辨率检测病毒基因中同义约束过剩区域的方法,核心科学问题是如何利用深度序列比对数据,通过严谨的统计模型检测短基因组中的重叠功能元件,技术路线为“方法开发→模拟数据验证→已知病毒元件验证→多样病毒新元件预测”的完整闭环。
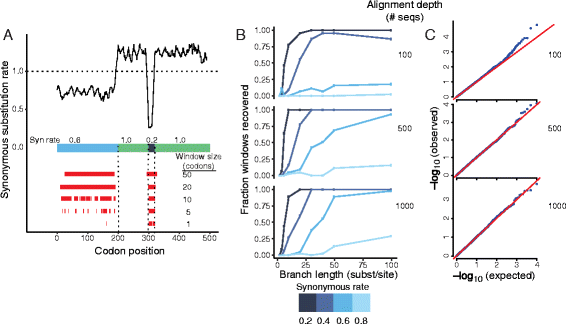
3.1 FRESCo方法开发与模拟数据验证
实验目的是构建基于系统发育密码子模型的同义约束区域检测方法,并验证其特异性与准确性。方法细节:采用HYPHY批处理语言实现FRESCo,先通过最大似然法拟合全局密码子模型参数(包括分支长度、密码子替换矩阵),再以滑动窗口方式,对每个窗口分别拟合允许局部同义替换率变化的替代模型与固定同义替换率的零模型,通过似然比检验(自由度1的卡方分布)判断约束区域的显著性;模拟不同比对深度(100、500、1000条序列)、约束强度(同义替换率为非约束区的20%-80%)、分支长度(2-100替换/位点)的序列数据进行方法验证。结果解读:模拟数据显示,FRESCo可准确恢复长而弱、短而强的同义约束区域,在Bonferroni校正后无假阳性结果;随着比对深度增加、约束强度增强、分支长度延长,约束区域的恢复率显著提升,如当分支长度从20替换/位点增加至40替换/位点、同义替换率为非约束区60%时,500条序列比对的恢复率从不足10%提升至50%以上;无约束模拟数据的P值符合均匀分布,表明统计检验的准确性。
实验所用关键产品:HYPHY批处理脚本、Muscle多序列比对工具、RAxML进化树构建工具、RNAz RNA二级结构预测工具、VARNA RNA结构可视化工具。
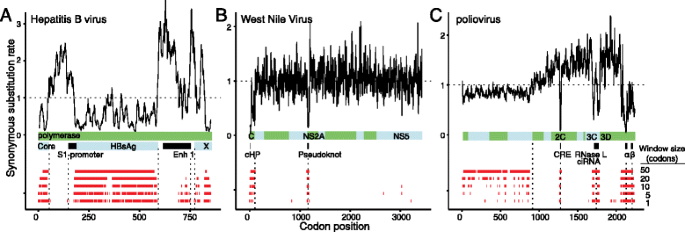
3.2 已知病毒基因组的功能元件验证
实验目的是验证FRESCo在真实病毒基因组中检测已知重叠功能元件的能力。方法细节:选取乙肝病毒(HBV)、西尼罗河病毒(WNV)、脊髓灰质炎病毒三种研究充分的病毒,从NCBI下载2000余株HBV、600余株WNV、300余株脊髓灰质炎病毒的编码序列,经Muscle进行氨基酸序列比对并转换为密码子比对,使用RAxML构建系统发育树后,应用FRESCo检测同义约束区域。结果解读:在HBV聚合酶基因中,检测到的同义约束区域均位于已注释的重叠功能区域,包括重叠的核心、HbsAg、X阅读框及增强子1、pre-S1启动子元件;在WNV中成功恢复衣壳编码区发夹(cHP)元件和NS2A基因中的假结元件,且衣壳基因全序列均存在同义约束信号,提示非cHP区的同义突变也可能影响病毒适应性;在脊髓灰质炎病毒中,恢复了2C基因中的顺式作用复制元件(CRE)、3C基因中的RNAse L抑制RNA、3D基因中的α和β元件,约束区域的同义替换率降至全基因组平均的10%-35%,同时发现结构区整体同义约束水平高于非结构区。
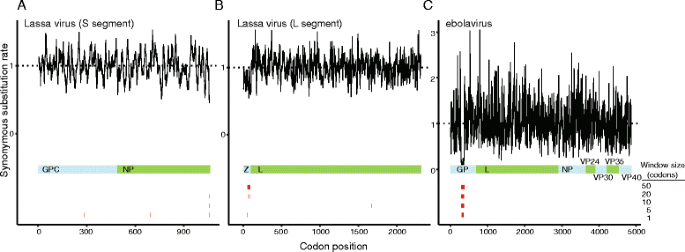
3.3 多样病毒基因组的新功能元件预测
实验目的是将FRESCo应用于多种不同类型的病毒,预测新的重叠功能元件。方法细节:选取30种具有不同基因组类型(DNA/RNA、单链/双链、分节段/不分节段)的病毒,从NCBI下载编码序列并完成比对与进化树构建,应用FRESCo检测同义约束区域,并通过RNAz预测区域的保守RNA二级结构以辅助功能推断。结果解读:在轮状病毒中,检测到NSP4、VP2、VP6基因末端的同义约束区域,补充了之前研究未报道的约束位点;在蓝舌病病毒NS3基因中发现内部同义约束区域,对应保守的+1阅读框或潜在RNA结构;在马铃薯Y病毒、芜菁花叶病毒的衣壳基因中检测到同义约束区域,且存在保守RNA二级结构,提示可能参与病毒复制调控;在埃博拉病毒GP基因中检测到对应RNA编辑位点的同义约束区域,拉沙病毒Z基因和NP基因末端发现新的同义约束区域,其中NP基因末端区域为回文结构,推测可能是RNA结合蛋白的作用位点。
4. Biomarker研究及发现成果
本研究中的Biomarker为同义约束元件(SCEs),是病毒基因组中具有功能意义的进化约束区域,可作为病毒功能注释、药物靶点筛选及疫苗设计的核心生物标志物。
同义约束元件的定位逻辑为“病毒编码序列比对→系统发育树构建→滑动窗口模型比较→似然比检验筛选显著约束区域”,验证逻辑为“模拟数据验证特异性→已知病毒元件验证准确性→多样病毒预测新元件→RNA结构预测辅助功能推断”。同义约束元件来源于30种病毒的临床分离株编码序列,验证方法为基于系统发育密码子模型的似然比检验,特异性在模拟数据中表现为Bonferroni校正后无假阳性,敏感性随比对深度、约束强度、分支长度提升,如1000条序列比对、分支长度100替换/位点、同义替换率为非约束区20%时,约束区域的恢复率接近100%;在真实病毒中,乙肝病毒的同义约束元件与已知重叠元件完全重合,验证了检测的准确性。
核心成果方面,同义约束元件与病毒的复制、包装、调控等关键生物学过程直接相关,如脊髓灰质炎病毒的CRE元件参与病毒RNA复制,埃博拉病毒的同义约束元件对应RNA编辑后的重叠阅读框,其氨基酸序列具有功能重要性;本研究的创新性在于首次开发了通用的高分辨率同义约束元件检测工具,在30种病毒中预测了大量未报道的重叠功能元件;统计学结果显示,所有显著同义约束元件的P值均小于1e-5(Bonferroni校正后P<0.05),部分病毒的检测样本量达数千株(如乙肝病毒n=2000+)。此外,同义约束元件可指导减毒活疫苗的开发,通过引入进化上被规避的同义突变降低病毒适应性,为抗病毒策略提供了新的靶点与思路。