1. 领域背景与文献引入
文献英文标题:Multi-species integrative biclustering;发表期刊:Genome Biology;影响因子:10.309(2010年);研究领域:计算系统生物学、微生物基因组学
随着基因组学数据的爆炸式增长,系统生物学领域已实现单生物体内全局调控网络推断和基因功能预测,但跨物种的调控模块保守性研究仍存在方法学瓶颈。传统聚类方法(如层次聚类、k-means)要求基因属于互斥簇,无法捕捉基因在不同条件下的多状态共调控;单物种双聚类方法虽能同时聚类基因和条件,但局限于单个物种,无法利用跨物种的进化信息。现有多物种分析方法分为两类:一类需匹配不同物种的实验条件,仅适用于有限的数据集;另一类基于共表达度量比较物种间差异,但多为顺序分析不同物种,且仅考虑表达数据,缺乏真正的多物种整合双聚类能力,也无法识别保守模块的物种特异性修饰。针对这些问题,本研究开发了多物种cMonkey算法,实现多物种多数据类型(表达、序列、网络)的整合双聚类,旨在识别微生物间的保守调控模块及物种特异性修饰,为理解细菌调控网络的进化提供新方法。
2. 文献综述解析
作者对领域内现有研究按分析维度分为单物种聚类、单物种双聚类、多物种聚类/双聚类三类。单物种聚类方法如层次凝聚聚类、k-means聚类,通过将基因分配到互斥簇中分析共表达模式,但无法捕捉基因在不同条件下的动态共调控,难以反映转录调控网络的复杂性。单物种双聚类方法如cMonkey、QUBIC,能同时聚类基因和实验条件,识别条件依赖的共表达模块,但仅适用于单个物种,无法利用跨物种的进化约束提升模块的生物学显著性。多物种分析方法则分为两类:一类要求匹配不同物种的实验条件,仅能分析具有直接对应条件的数据集,适用性受限;另一类基于共表达度量(如Pearson相关系数)比较物种间的共表达模式,如多物种k-means,但这类方法要么是顺序分析不同物种,要么仅考虑表达数据,缺乏真正的多物种整合双聚类能力,也无法整合序列、网络等多类型数据,更难以区分保守模块的核心部分和物种特异性修饰。
通过对比现有研究的局限性,本研究的创新价值凸显:首次提出真正的多物种整合双聚类算法,无需匹配实验条件即可整合多物种多数据类型进行分析,允许基因属于多个簇,同时能识别保守调控模块的核心部分和物种特异性修饰;算法通过逻辑回归平衡不同物种的得分权重,避免了数据量较大的物种对分析结果的主导,解决了现有方法在跨物种调控模块分析中的核心瓶颈。
3. 研究思路总结与详细解析
本研究的核心目标是开发多物种整合双聚类算法,用于识别微生物间的保守转录调控模块及物种特异性修饰;核心科学问题是如何在不依赖实验条件匹配的前提下,整合多物种多数据类型进行双聚类,同时平衡不同物种数据量的差异;技术路线遵循“算法开发→性能验证→生物学应用”的闭环:首先构建多物种cMonkey算法,包含直系同源基因识别、共享空间双聚类优化、物种特异性修饰优化三个核心步骤;然后通过与多种单/多物种聚类/双聚类方法的基准测试,验证算法的性能;最后将算法应用于枯草芽孢杆菌、炭疽芽孢杆菌、单核细胞增生李斯特菌的转录组数据,分析孢子形成和鞭毛组装的保守模块,揭示进化和功能差异。
3.1 多物种cMonkey算法开发
实验目的:构建可整合多物种多数据类型的双聚类算法,实现保守调控模块与物种特异性修饰的同步识别。
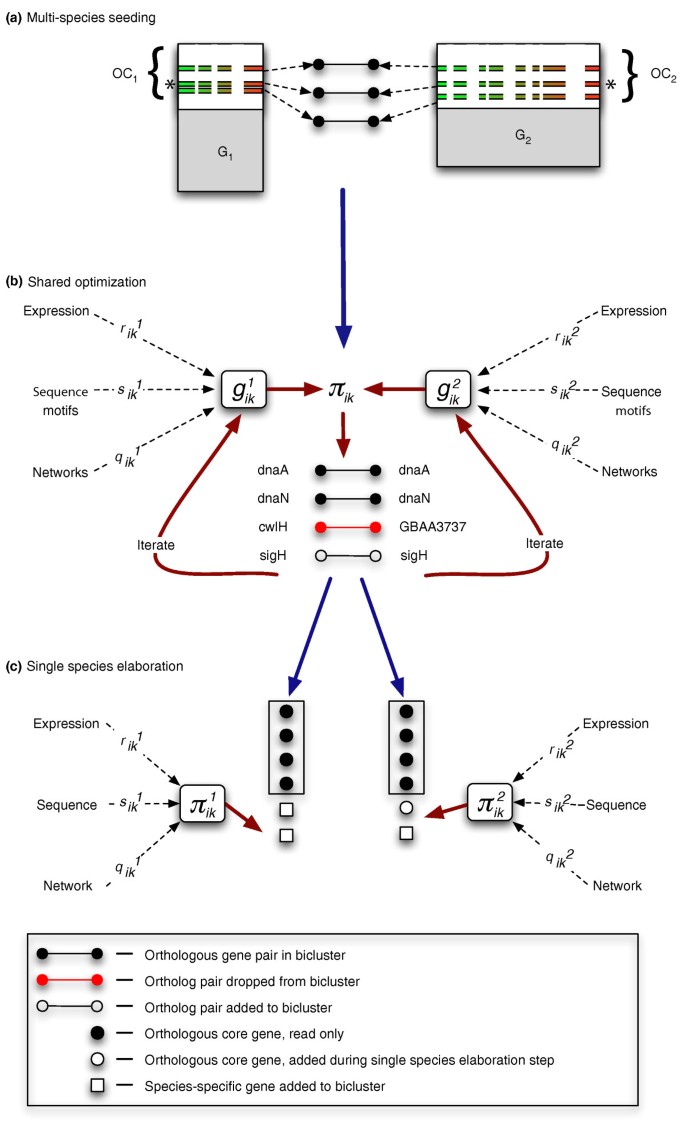
方法细节:算法分为四个核心步骤:1)直系同源基因识别:采用InParanoid算法识别物种间的直系同源基因对,支持一对多的同源关系;2)共享空间双聚类优化:以半随机方式生成种子,即随机选择一对直系同源基因,筛选该基因对在每个物种中差异表达最显著的70%实验条件,再添加5-10个与该基因对相关性最高的同源对;通过Monte Carlo模拟退火优化多物种多数据得分,迭代添加或移除同源对直至收敛;3)物种特异性修饰优化:将共享空间的双聚类拆分为单个物种的簇,固定保守核心基因,基于单物种数据集添加物种特异性基因;4)可选步骤:对未纳入保守模块的基因,使用单物种cMonkey算法识别物种特异性模块。算法整合表达数据、上游序列、代谢/信号通路网络等多类型数据,通过逻辑回归组合不同物种的得分,设置权重使各数据类型对聚类结果的影响均衡。
结果解读:算法可生成包含保守核心和物种特异性修饰的双聚类,保守核心由多物种共调控的同源基因组成,物种特异性修饰则补充了仅在单个物种中与核心模块共调控的基因,为后续的生物学分析提供了基础。
产品关联:文献未提及具体实验产品,领域常规使用R语言、MEME基序分析工具、InParanoid直系同源识别工具等。
3.2 算法性能基准测试
实验目的:验证多物种cMonkey算法的性能,与现有单/多物种聚类/双聚类方法进行全面比较。
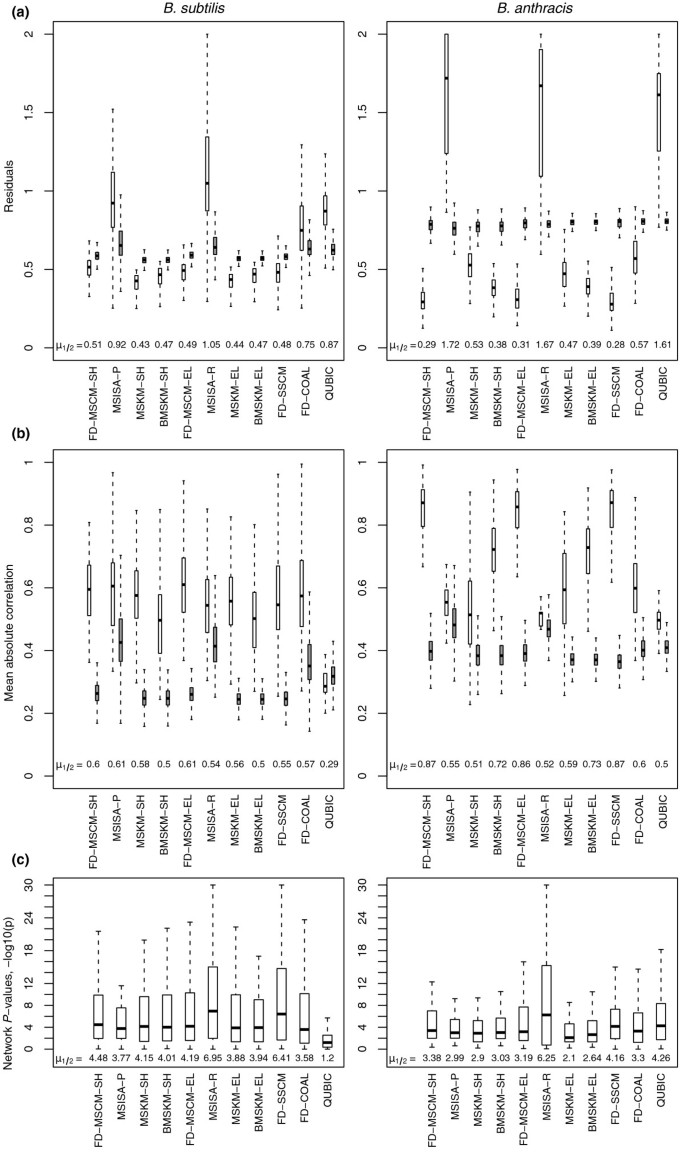
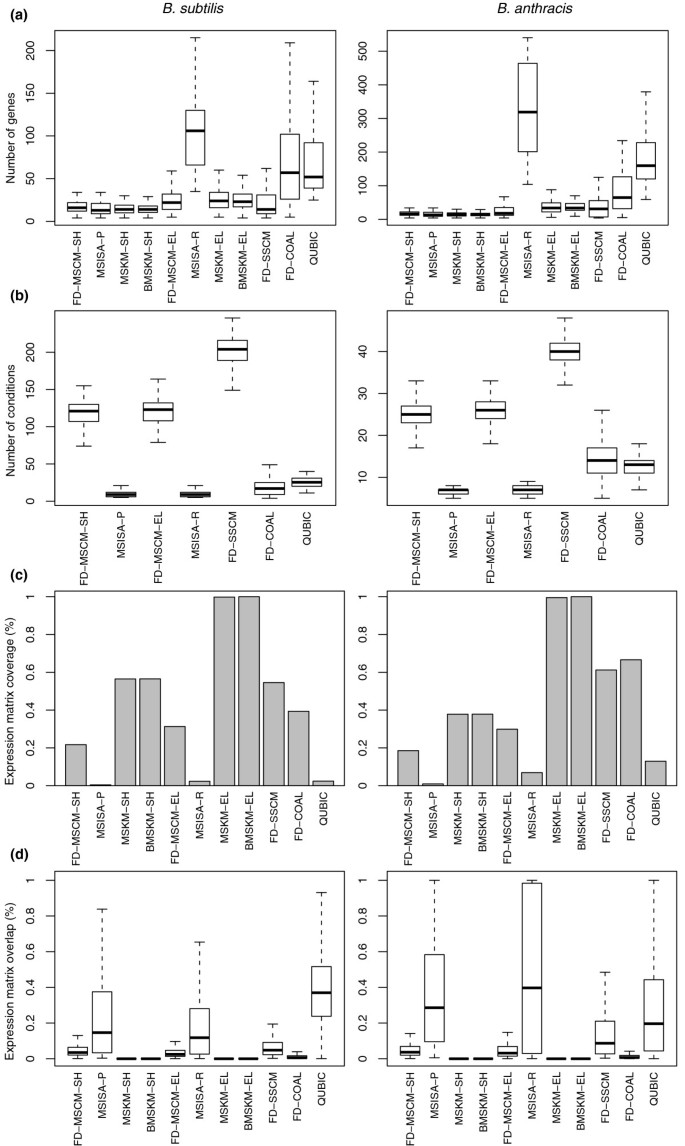
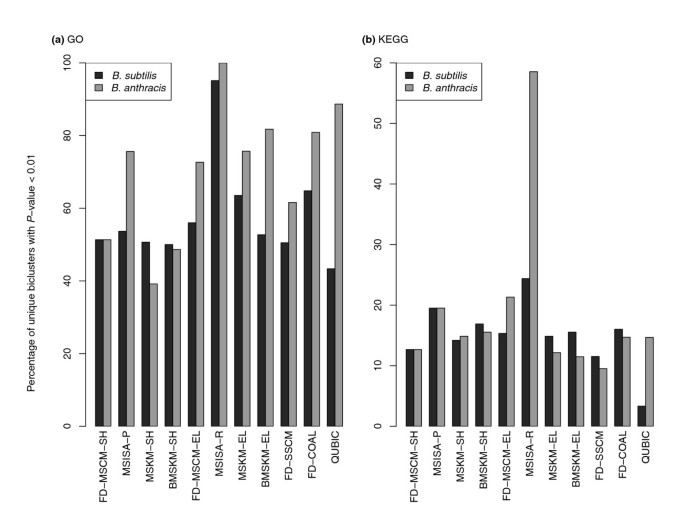
方法细节:选择枯草芽孢杆菌、炭疽芽孢杆菌、单核细胞增生李斯特菌的所有两两组合作为研究对象,比较的方法包括单物种方法(单物种cMonkey、QUBIC、Coalesce)和多物种方法(多物种迭代签名算法MSISA、多物种k-means MSKM、平衡多物种k-means BMSKM)。评估指标分为五类:1)双聚类一致性:包括表达残差、平均绝对相关性、网络关联P值、基序E值、序列P值;2)功能富集:双聚类的GO和KEGG通路富集比例;3)覆盖度:双聚类对表达数据矩阵的覆盖比例;4)重叠度:双聚类之间的重叠比例;5)保守性得分:基于F统计量计算的跨物种模块保守性。
结果解读:多物种cMonkey(MSCM)在所有五个一致性指标上表现优于或等于其他方法,其中在71/92的表达残差比较、92/92的平均相关性比较中表现最优;功能富集方面,添加物种特异性基因后,MSCM的GO富集双聚类比例从51.3%提升至56.0%(枯草芽孢杆菌),KEGG富集比例从12.7%提升至15.3%;保守性得分方面,MSCM的保守性得分>0.85,远高于单物种方法的<0.125,且避免了MSKM/BMSKM方法的过度保守问题;覆盖度和重叠度方面,MSCM的结果处于合理范围,既避免了QUBIC等方法的低覆盖度,也避免了MSISA的高重叠度。
3.3 保守调控模块的生物学分析
实验目的:应用多物种cMonkey算法分析微生物的保守调控模块,揭示跨物种的进化和功能差异。
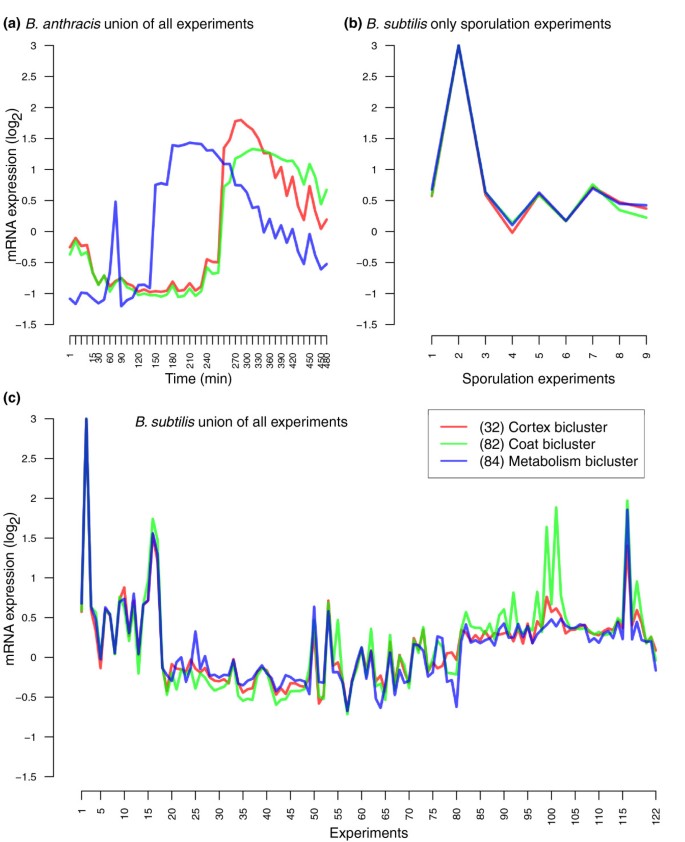
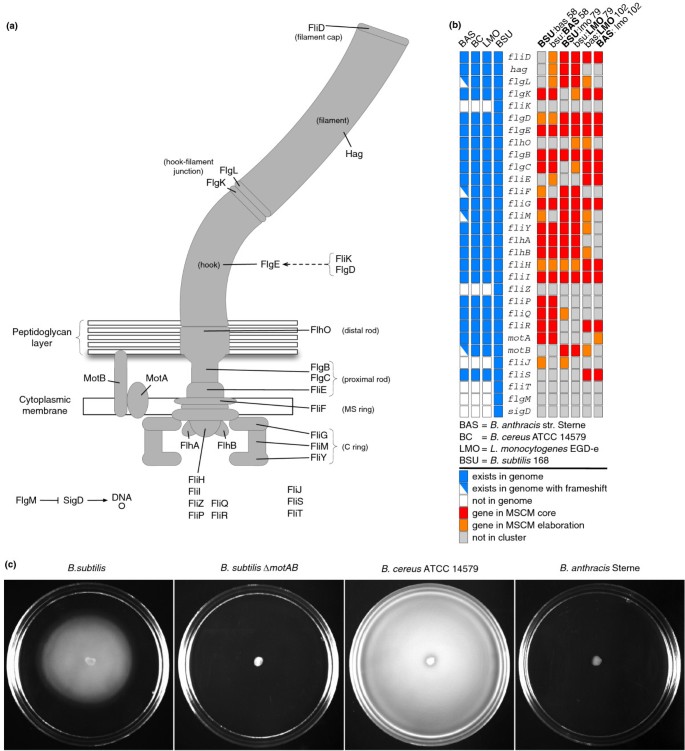
方法细节:1)孢子形成模块分析:针对枯草芽孢杆菌和炭疽芽孢杆菌的孢子形成相关双聚类,比较基因表达的时序模式,通过序列比对分析上游调控序列的差异;2)鞭毛组装模块分析:分析三个物种的鞭毛相关双聚类,对炭疽芽孢杆菌进行游泳运动实验(0.3%琼脂平板,30℃培养20小时)验证其运动性,通过BLAST和序列比对分析鞭毛基因的突变情况。
结果解读:孢子形成模块中,发现炭疽芽孢杆菌的代谢相关保守模块表达峰值在培养180分钟时出现,比皮层和外壳模块的表达峰值早2小时,而枯草芽孢杆菌中无此时序差异;序列分析显示,炭疽芽孢杆菌代谢模块的基因上游缺乏明显的σⁿ结合位点,提示转录调控重排是导致时序差异的原因。鞭毛组装模块中,尽管炭疽芽孢杆菌被认为是不运动的,但MSCM识别到保守的鞭毛组装模块;运动实验确认炭疽芽孢杆菌在30℃下无运动性,序列分析发现其motB、fliM、fliF等关键鞭毛基因存在移码突变,导致蛋白截断,解释了其非运动性,同时说明保守模块在表型丢失后仍可在基因组中保留一段时间。
产品关联:实验所用关键产品:LB培养基、0.3%琼脂平板(用于游泳运动实验),BLAST序列比对工具、MEME基序分析工具等。
4. Biomarker研究及发现成果
Biomarker定位:本文中的Biomarker为微生物间保守的转录调控模块,筛选逻辑为通过多物种整合双聚类算法,从直系同源基因对中识别在多物种中保守共调控的基因模块,再通过物种特异性修饰优化补充物种特异性基因,最后通过功能富集分析和实验验证确认模块的生物学意义。
研究过程详述:模块来源于枯草芽孢杆菌、炭疽芽孢杆菌、单核细胞增生李斯特菌的基因组和转录组数据,验证方法包括功能富集分析(GO、KEGG通路,P<0.01)、表达时序分析、游泳运动实验、序列突变分析。保守性方面,MSCM识别的模块保守性得分>0.85,显著高于单物种方法的<0.125;功能特异性方面,孢子形成模块的基因主要参与孢子发育的各个阶段,鞭毛组装模块的基因则集中于鞭毛的生物合成过程,功能富集P值均<0.01。
核心成果提炼:首次在非运动的炭疽芽孢杆菌中识别到保守的鞭毛组装模块,发现其关键基因存在移码突变,揭示了保守模块在表型丢失后的持续存在现象;发现枯草芽孢杆菌与炭疽芽孢杆菌孢子形成模块的表达时序差异,提示转录调控重排是细菌进化的重要机制;这些保守模块可作为微生物进化和功能研究的分子标志物,为理解细菌转录调控网络的进化提供了新视角。无临床Biomarker相关的敏感性、特异性数据,因本研究聚焦于基础研究层面的调控模块分析。