1. 领域背景与文献引入
文献英文标题:Generation of a spontaneous murine HPV + oral cancer model with site-specific oncogene insertion using CRISPR-SONIC;发表期刊:Cell & Bioscience;影响因子:未公开;研究领域:HPV相关头颈部肿瘤(口腔癌)模型构建与免疫治疗研究。
人乳头瘤病毒(HPV)是全球最常见的性传播感染病原体,约80%人群一生中会发生感染,高危型HPV(如16、18型)持续感染是宫颈癌、头颈部癌等恶性肿瘤的核心诱因。近年来,HPV相关头颈部癌(HPV+HNC)全球发病率显著上升,在高收入国家已超过宫颈癌成为最常见的HPV相关恶性肿瘤。现有预防性HPV疫苗可降低新发感染率,但无法清除已存在的感染,老年人群HPV+HNC的发病率仍在持续增长,预计到2045年70-83岁人群口咽癌发病率将从16.8/10万升至29.0/10万。临床现有治疗手段以手术、放化疗为主,严重影响患者生活质量,因此亟需开发新的治疗策略及精准的临床前模型。
当前HPV+HNC临床前模型存在诸多缺陷:异种移植模型依赖免疫缺陷小鼠,无法模拟肿瘤免疫微环境;转基因小鼠因胚胎期癌基因表达导致免疫耐受,且癌基因组织广泛表达不符合自然感染特征;条件性诱导模型构建耗时耗力,难以模拟HPV整合位点的多样性;转座子系统虽能快速成瘤,但整合位点半随机,无法模拟HPV精准整合的致癌效应。针对上述研究空白,本研究利用CRISPR-SONIC系统构建了HPV癌基因精准整合的自发性口腔癌小鼠模型,为HPV+HNC的机制研究与免疫治疗测试提供了全新平台。
2. 文献综述解析
作者按模型构建技术将现有HPV相关肿瘤模型分为四类:异种移植模型、转基因模型、条件性诱导模型、转座子介导的随机整合模型,系统评述了各类模型的优势与局限性,明确了精准整合模型的核心研究需求。
异种移植模型通过将HPV阳性肿瘤细胞系植入免疫缺陷小鼠体内,可快速诱导肿瘤形成,但细胞系在培养过程中发生适应性改变,且免疫缺陷环境无法模拟临床肿瘤的免疫微环境,难以用于免疫治疗测试。转基因模型通过胚胎期插入HPV癌基因获得免疫健全小鼠,但胚胎期癌基因持续表达会导致宿主对HPV抗原产生免疫耐受,同时癌基因组织广泛表达与自然感染的局灶性特征不符,影响治疗测试准确性。条件性诱导模型利用Cre重组酶实现癌基因时空特异性表达,解决了组织广泛表达的问题,但模型构建需复杂的基因修饰与繁育过程,耗时且成本高,无法高效探索HPV整合位点的多样性。转座子系统(如Sleeping Beauty)通过半随机整合癌基因可快速诱导自发肿瘤,且模型具有免疫健全性,但整合位点随机性无法模拟HPV精准整合的致癌效应,难以研究整合位点对肿瘤发生发展的影响。
本研究的创新价值在于首次将CRISPR-SONIC系统应用于HPV相关口腔癌模型构建,实现了HPV16 E6/E7癌基因在宿主β-actin 3’UTR位点的精准整合,同时可在免疫健全或免疫缺陷小鼠中诱导自发肿瘤,既解决了传统模型免疫环境不真实的问题,又能模拟HPV精准整合的致癌过程,为研究HPV整合的致癌机制和测试免疫治疗策略提供了更精准的平台。
3. 研究思路总结与详细解析
本研究的整体目标是构建基于CRISPR-SONIC系统的精准整合HPV16 E6/E7的小鼠口腔癌模型,验证其成瘤能力及对HPV特异性免疫治疗的响应性,同时构建不同癌基因组合的模型以模拟临床不同分子亚型的HPV+HNC。核心科学问题包括:精准整合的HPV癌基因能否在小鼠体内诱导自发肿瘤?该模型能否模拟临床HPV+HNC对免疫治疗的响应?不同癌基因组合对肿瘤形态和分子特征的影响是什么?技术路线遵循“载体构建→体外验证→体内成瘤→免疫治疗测试→模型优化”的闭环逻辑,逐步验证模型的有效性和临床相关性。
3.1 载体构建与体外功能验证
实验目的:验证CRISPR-SONIC系统能否在细胞中实现HPV16 E6/E7及癌基因的精准整合与稳定表达。方法细节:构建包含KrasG12D、荧光素酶(Luc)、HPV16 E7、E6的SONIC供体载体SONIC-KrasG12D-Luc-E7-E6,将该载体与靶向小鼠β-actin 3’UTR的sgRNA质粒、Cas9质粒以1:1:1的比例共转染TC-1细胞,同时设置转座子系统(Sleeping Beauty)作为阳性对照,以及缺失关键整合元件的对照组。转染后3天和7天,通过IVIS Spectrum成像系统检测荧光素酶的生物发光信号,验证基因的整合与表达。结果解读:转染后3天,CRISPR-SONIC组与对照组的生物发光信号无显著差异;转染后7天,CRISPR-SONIC组的生物发光信号显著高于对照组(P<0.0001,n=3),说明CRISPR-SONIC系统实现了癌基因的稳定整合与表达;转座子系统组在转染后3天即呈现较高的生物发光信号,但其对照组也存在一定的基础表达,提示转座子系统的表达水平更高但整合特异性较低。
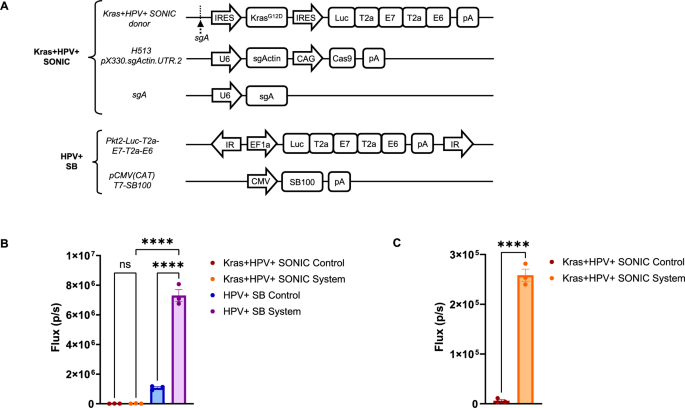
产品关联:实验所用关键产品:ExpiFectamine 293 Reagent(Gibco)、IVIS Spectrum imager series 2000(Xenogen)、D-luciferin(GoldBio)、Living Image软件(Xenogen)等。
3.2 免疫健全小鼠中肿瘤诱导与免疫抑制必要性验证
实验目的:验证CRISPR-SONIC系统能否在免疫健全小鼠中诱导自发口腔肿瘤,明确免疫抑制对成瘤的必要性。方法细节:将C57BL/6小鼠分为两组,每组n=5,一组通过腹腔注射抗CD3抗体(连续3天)进行短暂免疫抑制,另一组不进行免疫抑制处理。在免疫抑制组的第3天,将SONIC载体、sgRNA质粒、Cas9质粒混合后经颊黏膜下注射并电穿孔,每周通过生物发光成像监测肿瘤生长情况,同时设置转座子系统组作为对照。结果解读:短暂免疫抑制组的小鼠颊部肿瘤生物发光信号随时间持续升高,表明成功诱导自发肿瘤;而未免疫抑制的免疫健全小鼠,肿瘤生物发光信号先升高后快速下降,提示宿主免疫系统清除了肿瘤细胞,证明短暂免疫抑制是CRISPR-SONIC系统诱导自发肿瘤的必要条件。与转座子系统组相比,CRISPR-SONIC组的小鼠存活率更高(文献未明确具体数值,基于图表趋势推测),说明该模型的成瘤过程更可控。
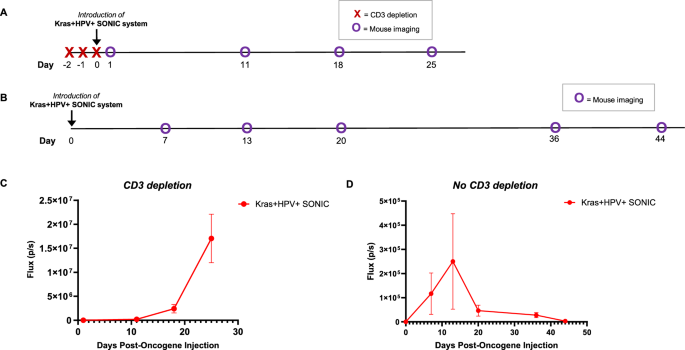
产品关联:实验所用关键产品:抗CD3抗体(Bio X Cell,clone 17A2)、ECM 830 Square Wave Electroporation System(BTX)等。
3.3 治疗性HPV DNA疫苗对肿瘤的控制作用验证
实验目的:验证CRISPR-SONIC诱导的HPV阳性肿瘤能否对HPV特异性免疫治疗产生响应。方法细节:在经抗CD3短暂免疫抑制的C57BL/6小鼠中诱导颊部肿瘤后,一组小鼠接受pNGVL4a-CRT/E7(detox) DNA疫苗肌肉注射并电穿孔,共进行4次基础免疫和1次加强免疫,另一组作为未疫苗对照组。每周通过生物发光成像监测肿瘤生长,在免疫后第39天采集外周血,通过流式细胞术检测E7特异性CD8+T细胞的比例。结果解读:未疫苗对照组的肿瘤生物发光信号持续升高,而疫苗组的信号逐渐降至基线水平,表明治疗性疫苗有效控制了肿瘤生长;流式细胞术结果显示,疫苗组外周血中E7特异性CD8+T细胞占总CD8+T细胞的比例显著高于未疫苗组(P<0.01,n=5),证明疫苗诱导了特异性的细胞免疫反应,且该反应与肿瘤控制直接相关。
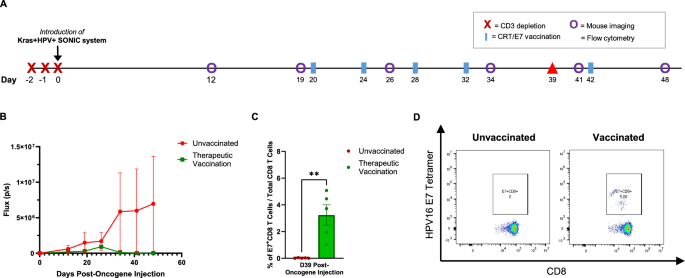
产品关联:实验所用关键产品:pNGVL4a-CRT/E7(detox) DNA疫苗、Zombie Aqua Live/Dead染色剂(BioLegend)、Fc block(BD Pharmingen)、APC/Fire™750 anti-mouse CD8a抗体(BioLegend)、PE标记的HPV16 E7aa49-57肽负载H-2Db四聚体(NIAID tetramer core facility)、CytoFLEX S流式细胞仪(Beckman Coulter)、FlowJo软件(BD Biosciences)等。
3.4 CD4+T细胞缺陷环境中免疫治疗的响应性验证
实验目的:模拟HIV感染等CD4+T细胞缺陷的临床场景,验证HPV DNA疫苗在免疫缺陷宿主中的治疗与预防效果。方法细节:将C57BL/6小鼠分为两组,每组n=5,通过腹腔注射抗CD4抗体(连续3天初始清除,之后每周维持清除)构建CD4+T细胞缺陷模型。一组小鼠在肿瘤诱导后接受治疗性疫苗接种,另一组在肿瘤诱导前接受预防性疫苗接种,同时设置未疫苗对照组。通过生物发光成像监测肿瘤生长,采集外周血检测E7特异性CD8+T细胞比例。结果解读:治疗性疫苗组的肿瘤生长显著被抑制,预防性疫苗组的肿瘤生物发光信号显著低于未疫苗组(P<0.05,n=5);流式细胞术结果显示,无论是治疗性还是预防性疫苗组,E7特异性CD8+T细胞的比例均显著高于未疫苗组,证明疫苗可在CD4+T细胞缺陷的环境中诱导有效的E7特异性细胞免疫反应,实现肿瘤控制或预防。
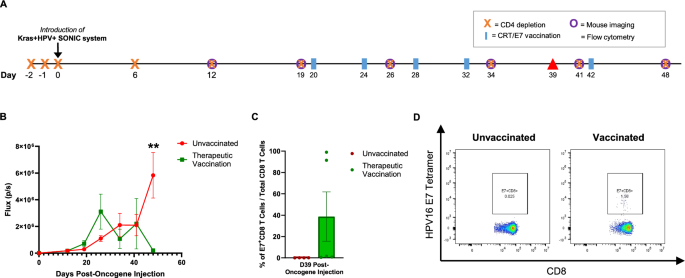
产品关联:实验所用关键产品:抗CD4抗体(Bio X Cell,clone GK1.5)等。
3.5 临床相关癌形态模型构建与分子特征验证
实验目的:构建更接近临床HPV+HNC形态特征的模型,模拟PI3K/AKT/mTOR通路激活的分子亚型。方法细节:在NSG免疫缺陷小鼠(n=3)的颊黏膜下注射包含HPV16 E6/E7的SONIC载体、sgRNA质粒、Cas9质粒,同时联合SB转座子系统递送AKT和c-Myc癌基因,通过电穿孔促进基因整合。待肿瘤形成后,进行组织学(苏木精-伊红染色,H&E)和免疫组化分析,验证肿瘤形态与分子特征。结果解读:H&E染色显示肿瘤呈现癌的典型形态特征,包括高核质比、核异型性、频繁核分裂象、坏死与出血;免疫组化结果显示肿瘤细胞表达上皮标志物CK19和OSCAR,增殖标志物Ki67高表达,同时c-Myc和AKT呈特异性表达(核和膜定位),证明该模型模拟了临床HPV+HNC的癌形态和分子特征;而KrasG12D组合的模型呈现肉瘤形态,说明不同癌基因组合可诱导不同组织学类型的肿瘤,为构建多样化的HPV+HNC模型提供了依据。
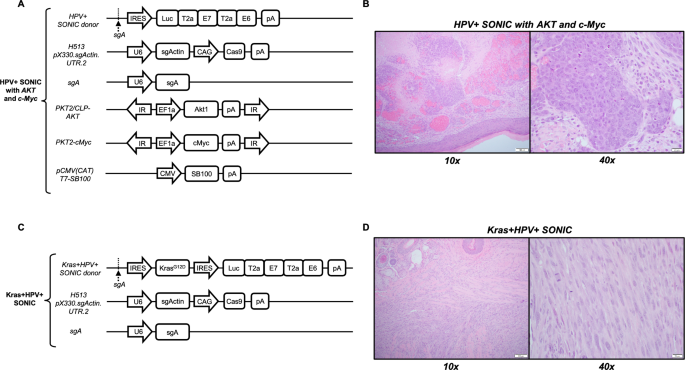
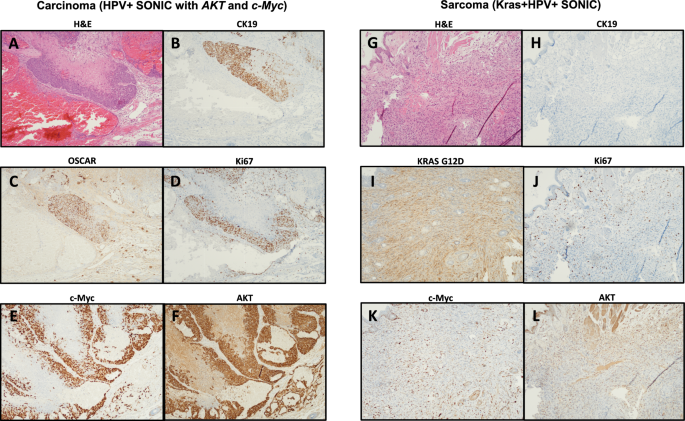
产品关联:实验所用关键产品:免疫组化抗体(Abcam anti-cMyc、Proteintech anti-AKT、Abcam anti-Ki67、Abcam anti-CK19、ThermoFischer anti-KRAS-G12D等)、Ventana Discovery Ultra自动染色仪(Roche Diagnostics)等。
4. Biomarker研究及发现成果解析
本文涉及的Biomarker包括功能性免疫Biomarker(E7特异性CD8+T细胞)和肿瘤分子特征Biomarker(HPV16 E6/E7、KrasG12D、AKT、c-Myc),通过体外验证、体内成瘤、免疫治疗测试等环节完成筛选与验证,为HPV相关肿瘤的免疫治疗响应预测和模型分子特征鉴定提供了依据。
E7特异性CD8+T细胞作为免疫治疗响应的功能性Biomarker,筛选逻辑是基于HPV DNA疫苗靶向E7抗原,通过流式细胞术检测其在免疫治疗前后的变化,验证其与肿瘤控制的相关性;肿瘤分子特征Biomarker的验证逻辑是通过PCR和免疫组化检测其在肿瘤组织中的表达,确认模型的分子特征与临床亚型的一致性。
E7特异性CD8+T细胞的来源为小鼠外周血单个核细胞(PBMCs),验证方法为流式细胞术,采用E7aa49-57肽负载的H-2Db四聚体进行特异性染色,其特异性表现为仅在疫苗免疫的小鼠中显著升高,敏感性为可检测到低至0.1%的特异性T细胞比例;在治疗性疫苗组中,E7特异性CD8+T细胞占总CD8+T细胞的比例约为1.5%,显著高于未疫苗组的0.2%(P<0.01,n=5)。肿瘤组织中HPV16 E6/E7的验证方法为RT-PCR,所有CRISPR-SONIC诱导的肿瘤均检测到E6/E7的表达(阳性率100%,n=5);KrasG12D、AKT、c-Myc的验证方法为免疫组化,对应组合的肿瘤中均呈阳性表达(阳性率100%,n=3/5)。
核心成果提炼:E7特异性CD8+T细胞可作为HPV相关肿瘤免疫治疗响应的预测Biomarker,其比例升高与肿瘤控制直接相关,在治疗性疫苗组中,肿瘤控制率达100%(n=5);首次在精准整合模型中证明,不同癌基因组合可诱导不同组织学类型的肿瘤,KrasG12D组合诱导肉瘤形态,AKT+c-Myc组合诱导临床相关的癌形态,为构建不同分子亚型的HPV+HNC模型提供了实验依据;该模型中HPV16 E6/E7的精准整合,为研究HPV整合位点对致癌效应的影响提供了平台,有助于揭示HPV整合的分子机制。统计学结果:E7特异性CD8+T细胞比例在疫苗组 vs 未疫苗组,P<0.01(n=5);治疗性疫苗组 vs 未疫苗组的肿瘤生长曲线,P<0.01(n=5);预防性疫苗组 vs 未疫苗组的肿瘤生长曲线,P<0.05(n=5)。