1. 领域背景与文献引入
文献英文标题:Online biophysical predictions for SARS-CoV-2 proteins;发表期刊:BMC Molecular and Cell Biology;影响因子:未公开;研究领域:冠状病毒分子生物学(SARS-CoV-2蛋白功能与生物物理特征研究)
COVID-19全球大流行以来,SARS-CoV-2的分子生物学与结构生物学研究成为生命科学领域的核心热点。领域发展关键节点包括2020年初科研团队成功解析刺突蛋白三维结构,推动了mRNA疫苗与中和抗体药物的快速研发;后续多项研究陆续解析了病毒复制酶、核蛋白等关键蛋白的核心功能区域结构,为抗病毒药物靶点筛选提供了直接依据。当前研究热点聚焦于病毒蛋白的结构-功能关系、宿主-病毒相互作用机制及耐药突变的结构基础,但领域内仍存在未解决的核心问题:大量SARS-CoV-2蛋白或其功能区域缺乏高分辨率三维结构信息,静态结构生物学方法无法覆盖这些区域;同时,静态结构仅能反映蛋白的一种稳定状态,无法体现序列编码的固有生物物理倾向(如动态构象变化、无序区域的潜在功能),基于已知结构的分子动力学模拟也无法有效研究这些未折叠或动态变化的区域。
针对这一研究空白,本研究团队开发了一款基于蛋白序列的在线生物物理预测平台,通过整合多种成熟的生物信息学预测工具,系统分析27种SARS-CoV-2蛋白的固有生物物理特征,同时纳入同源蛋白的生物物理变异信息,为研究者探索未解析结构区域的潜在功能提供数据支持与假设依据,填补了静态结构研究与动态功能探索之间的缺口,具有推动SARS-CoV-2蛋白功能研究全面覆盖的学术价值。
2. 文献综述解析
本文综述部分以SARS-CoV-2蛋白结构生物学研究的现状与局限为核心评述逻辑,将现有研究分为“已解析三维结构的蛋白功能研究”与“未解析结构区域的功能未知问题”两大维度,系统梳理了领域研究的进展与不足。
现有结构生物学研究已成功解析SARS-CoV-2的刺突蛋白、复制酶、核蛋白等关键蛋白的核心功能区域三维结构,这些成果为理解病毒的感染机制、开发结构导向的抗病毒药物提供了直接依据,其技术优势在于能精准呈现蛋白的静态折叠状态与关键功能位点的空间分布。但现有研究存在明显局限性:一方面,仍有大量病毒蛋白或其功能区域缺乏高分辨率结构信息,无法通过结构生物学方法直接研究;另一方面,静态结构仅能反映蛋白的一种稳定状态,无法体现序列编码的固有生物物理倾向,而基于已知结构的分子动力学模拟也无法覆盖这些未折叠或动态变化的区域。
通过对比现有研究的不足,本研究的核心创新点在于首次将多种基于蛋白序列的生物物理预测工具进行整合,针对27种SARS-CoV-2蛋白提供了残基水平的多维度生物物理特征预测,包括主链与侧链动态、无序倾向、折叠起始概率、β-折叠聚集倾向、蛋白-蛋白相互作用位点及构象表位等;同时,研究还纳入了同源蛋白的生物物理变异分析,能反映病毒蛋白功能相关的生物物理行为的进化范围。这一研究突破了静态结构研究的局限,为研究者提供了探索未解析结构蛋白功能的新视角,其学术必要性在于能帮助研究者针对未知功能区域形成可验证的科学假设,推动SARS-CoV-2蛋白功能研究的全面覆盖。
3. 研究思路总结与详细解析
本研究的整体目标是开发一个用户友好的在线平台,提供SARS-CoV-2蛋白的多维度生物物理序列预测,核心科学问题是如何通过蛋白序列编码的信息揭示未解析结构区域的动态行为与潜在功能,技术路线遵循“数据构建→特征预测→可视化平台开发→案例验证”的闭环逻辑,确保研究结果的科学性与实用性。
3.1 数据集构建与多序列比对分析
实验目的是获取高质量的SARS-CoV-2蛋白序列及同源蛋白序列数据集,为后续生物物理特征预测提供序列基础与进化信息。方法细节为从UniProt数据库的COVID-19专区及NCBI的SARS-CoV-2资源平台获取27种病毒蛋白的目标序列;针对每个序列,通过UniProt BLAST工具在UniRef90数据库中搜索同源序列,限制命中数为250条;使用CD-HIT工具对同源序列进行去冗余处理,默认采用70%的序列同一性阈值,针对P0DTC2蛋白则调整为80%以避免丢失过多有效序列;将去冗余后的代表性序列与目标序列一起,通过基于Clustal Omega的UniProt在线比对工具进行多序列比对,并去除同源序列中超出目标序列的N端与C端区域;对于15个非结构蛋白,先以ORF1ab全长序列进行BLAST搜索、去冗余与比对,再拆分为单个非结构蛋白序列。结果解读显示,本研究成功构建了覆盖27种SARS-CoV-2蛋白的高质量多序列比对数据集,既保留了目标序列的完整性,又纳入了同源蛋白的进化信息,为后续的生物物理特征预测与变异分析提供了可靠基础。产品关联:文献未提及具体实验产品,领域常规使用UniProt BLAST、CD-HIT、Clustal Omega等开源生物信息学工具。
3.2 多维度生物物理特征的序列预测
实验目的是基于蛋白序列预测多种核心生物物理特征,揭示SARS-CoV-2蛋白的固有生物物理倾向,而非仅依赖于最终折叠状态。方法细节包括采用DynaMine工具预测残基水平的主链动态与侧链动态及构象倾向,该工具基于NMR化学位移数据训练的线性回归模型;使用EFoldMine工具预测残基的早期折叠概率,该工具以5残基片段的5种DynaMine特征构建25维特征向量,通过支持向量机(SVM)模型训练,训练集为30种蛋白的高分辨率氢-氘交换(HDX)NMR数据;采用DisoMine工具预测无序区域倾向,该工具基于循环神经网络(RNN)模型,输入特征包括DynaMine的动态数据、EFoldMine的折叠概率数据及PSIPRED的二级结构预测结果;使用Agmata工具预测β-折叠聚集倾向,该工具采用逻辑回归模型,基于3残基窗口的DynaMine特征构建统计势能模型;通过SeRenDIP工具预测蛋白-蛋白相互作用位点,该工具基于随机森林模型,训练集为PDB数据库中的同源与异源相互作用数据集;采用SeRenDIP-CE工具预测构象表位倾向,训练集为SabDab数据库中的抗体结合区域序列;同时,使用PSPer工具预测蛋白的FUS样相分离行为,该工具采用隐马尔可夫模型(HMM)识别低复杂度区域并评估相分离潜能。此外,对所有同源蛋白序列进行相同的特征预测,通过箱线图分析每个残基位置的生物物理特征变异范围。结果解读显示,本研究成功获得了27种SARS-CoV-2蛋白的残基水平多维度生物物理特征预测值,包括动态、折叠、无序、聚集、相互作用等,同时揭示了同源蛋白中这些特征的进化变异范围,能反映病毒蛋白的固有生物物理倾向,为探索未知功能区域提供了数据支持。产品关联:文献未提及具体实验产品,领域常规使用DynaMine、EFoldMine、DisoMine等开源生物信息学预测工具。
3.3 在线可视化平台的开发与功能实现
实验目的是开发一个用户友好的在线平台,将复杂的生物物理预测数据进行可视化展示,方便研究者查询与分析。方法细节为采用Django框架搭建网站后端,使用ApexCharts JavaScript库实现前端的交互式可视化;网站首页提供平台功能介绍与使用指引,“Entries”页面以可排序表格展示27种SARS-CoV-2蛋白的基本信息(ORF名称、RefSeq ID、UniProt ID、序列长度、蛋白类别);每个蛋白的详情页面提供多维度功能:链接到PDB数据库的结构信息(若有)、UniProt与NCBI的蛋白信息、PSPer相分离预测结果(仅针对序列长度≥130的蛋白)、预测数据的JSON格式下载、多序列比对文件下载;详情页面的顶部图表以蛋白序列为X轴,展示所有生物物理特征的预测值,支持点击图例切换不同特征的显示,鼠标悬停可查看具体残基的预测数值;第二个图表展示特定生物物理特征的同源蛋白变异范围,包括中位数、四分位数与异常值范围,同时叠加目标蛋白的预测值。结果解读显示,开发的https://bio2byte.be/sars2/在线平台实现了预测数据的直观可视化与便捷查询,研究者可通过交互式图表快速分析蛋白的生物物理特征,下载数据进行进一步的深入研究,平台的实用性已通过案例验证得到体现。产品关联:文献未提及具体实验产品,领域常规使用Django、ApexCharts等网站开发工具。
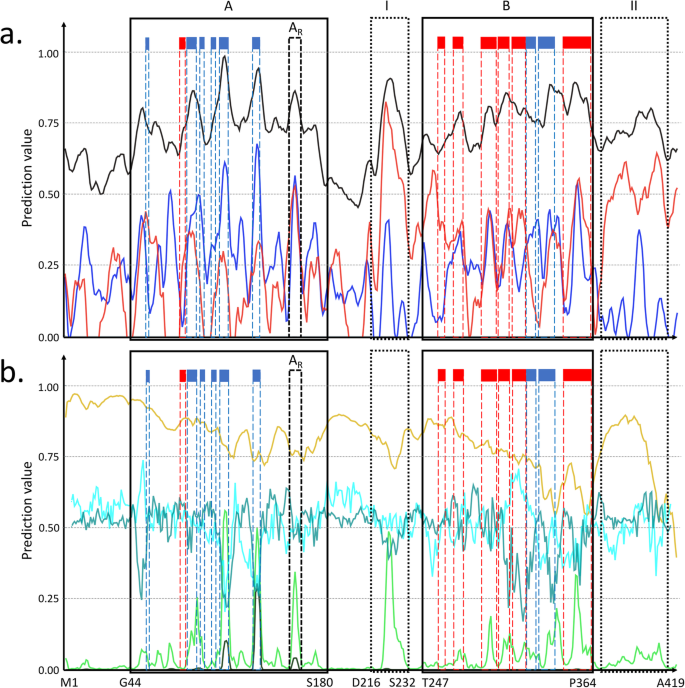
3.4 核蛋白P0DTC9的案例验证分析
实验目的是通过具体案例验证预测数据的实用性,展示如何利用生物物理预测结果探索未解析结构区域的潜在功能。方法细节为选取SARS-CoV-2核蛋白P0DTC9(序列长度419氨基酸)作为研究案例,该蛋白已有两个结构区域被解析:Gly44-Ser180(RNA结合域)与Thr247-Pro364(寡聚化域);结合已有的结构信息,分析预测的主链动态、无序倾向、折叠起始、蛋白-蛋白相互作用、表位倾向等特征,同时对比同源蛋白的生物物理变异范围。结果解读显示,预测结果与已解析结构高度一致:已折叠的结构区域对应主链动态值低(刚性高)、无序倾向低的区域;而N端Gly44之前的区域显示高灵活性、蛋白-蛋白相互作用倾向,且包含多个已验证的磷酸化位点(Ser23、Ser26),推测:该区域可能参与病毒复制的调控;Ser180-Thr247的连接区域中,Asp216-Thr247段显示高主链刚性与螺旋倾向,且早期折叠概率高于同源蛋白的第三四分位数范围,推测:该区域在SARS-CoV-2中具有更强的自主螺旋形成倾向,可能通过形成螺旋调控两个结构域之间的距离或结合位点;C端Pro364之后的区域显示高螺旋倾向、蛋白-蛋白相互作用与表位倾向,推测:该区域可能具有潜在的调控或免疫功能。这些预测结果为未解析结构区域的功能研究提供了可验证的科学假设,充分体现了平台的实用性。产品关联:文献未提及具体实验产品,领域常规使用PDB数据库获取蛋白结构信息。
4. Biomarker研究及发现成果
本研究虽未直接聚焦疾病诊断或预后Biomarker,但通过生物物理特征预测与磷酸化位点整合,挖掘了SARS-CoV-2蛋白中的功能相关Biomarker候选,包括调控型磷酸化位点与免疫型表位倾向区域,为病毒感染机制研究与疫苗靶点开发提供了重要线索。
Biomarker定位:本研究涉及的Biomarker类型包括两类:一是病毒蛋白的磷酸化位点(调控型功能Biomarker),二是构象表位倾向区域(免疫型Biomarker)。筛选与验证逻辑为:磷酸化位点通过整合PRIDE数据库中两个独立SARS-CoV-2磷酸化组学项目的数据集,筛选在多个项目中出现且定位概率>0.6的高可信度位点;构象表位倾向区域通过SeRenDIP-CE模型预测,该模型基于SabDab数据库中的抗体结合区域序列训练,能识别具有潜在抗体结合能力的蛋白区域。
研究过程详述:磷酸化位点的来源为PRIDE数据库中的PXD020183与PXD019113两个SARS-CoV-2磷酸化组学项目,验证方法为整合两个项目的数据分析,仅保留定位概率>0.6且在两个项目中均出现的位点;构象表位倾向区域通过SeRenDIP-CE工具的随机森林模型预测,模型训练集包含已标注的抗体结合区域序列。文献未提供这些Biomarker的特异性与敏感性数据。
核心成果提炼:研究发现核蛋白P0DTC9等多个SARS-CoV-2蛋白存在高可信度磷酸化位点,如核蛋白的Ser23、Ser26、Ser79、Ser187等,推测:这些位点可能参与病毒复制过程中的调控功能;预测的构象表位倾向区域为SARS-CoV-2疫苗的靶点设计提供了候选区域,尤其是未被结构生物学研究覆盖的蛋白区域。本研究的创新性在于首次通过多维度生物物理预测与组学数据整合,系统挖掘了SARS-CoV-2蛋白中的功能Biomarker候选,为病毒感染机制研究与免疫干预策略开发提供了新的方向;文献未明确提供相关统计学结果(如磷酸化位点的富集分析P值、表位预测的准确性数据)。