1. 领域背景与文献引入
文献英文标题:Post-translational modification of CDK1–STAT3 signaling by fisetin suppresses pancreatic cancer stem cell properties;发表期刊:Cell & Bioscience;影响因子:未明确提供;研究领域:胰腺癌干细胞与肿瘤耐药治疗。
胰腺癌导管腺癌(PDAC)是临床恶性程度最高的肿瘤之一,5年生存率仅9%,早期诊断率不足20%,绝大多数患者确诊时已处于晚期,失去手术根治机会。现有一线化疗药物如吉西他滨的治疗效果有限,化疗耐药是导致PDAC患者预后极差的核心原因。领域共识:癌症干细胞(CSCs)是肿瘤组织中具有自我更新能力和多向分化潜能的细胞亚群,是肿瘤发生、转移、复发及化疗耐药的关键驱动因素,靶向清除CSCs是突破PDAC治疗瓶颈的重要方向。
细胞周期蛋白依赖性激酶(CDK)家族在细胞周期调控中发挥核心作用,其异常激活可促进肿瘤细胞无限增殖。已有研究表明CDK1在黑色素瘤、膀胱癌等多种肿瘤中调控干细胞 pluripotency,但CDK1在PDAC干细胞中的功能及调控机制尚未阐明。非瑟酮是一种天然黄酮类化合物,广泛存在于多种蔬菜水果中,已被证实具有广谱抗肿瘤活性,可抑制多种肿瘤细胞增殖,但非瑟酮对PDAC干细胞的作用及分子靶点仍不明确。
针对PDAC干细胞靶向治疗的研究空白,以及CDK1在PDAC中功能的未知性,本研究聚焦CDK1-STAT3信号通路的翻译后修饰调控机制,旨在解析非瑟酮抑制PDAC干细胞特性的分子靶点,为PDAC耐药逆转提供新的治疗策略和理论依据。
2. 文献综述解析
作者从PDAC治疗困境出发,围绕CSCs耐药机制、CDK家族在肿瘤干细胞中的调控作用、天然化合物靶向CSCs的研究进展三个维度构建综述逻辑,系统梳理领域内现有研究的成果与局限性,明确本研究的创新方向。
现有研究已证实,PDAC的化疗耐药性与CSCs的自我更新能力密切相关,吉西他滨治疗可诱导PDAC细胞向CSCs表型转化,进一步增强耐药性;CDK家族成员如CDK4/6已成为肿瘤治疗的重要靶点,但CDK1在PDAC中的功能及调控通路尚未报道;非瑟酮可通过诱导DNA损伤、自噬等途径抑制PDAC细胞增殖,但对CSCs的作用机制未明确。现有研究的局限性在于,缺乏对CDK1在PDAC干细胞中调控网络的解析,以及非瑟酮靶向CSCs的具体分子机制研究,无法为临床联合治疗提供直接的理论支持。
本研究通过对比现有研究的空白,首次将CDK1的翻译后修饰与PDAC干细胞特性调控关联,明确非瑟酮通过靶向CDK1的激酶口袋结构域,诱导其K33位点乙酰化,进而抑制STAT3通路激活,填补了CDK1乙酰化调控胰腺癌干细胞的研究空白,为非瑟酮联合吉西他滨治疗PDAC提供了关键的机制依据,具有重要的学术价值和临床转化潜力。
3. 研究思路总结与详细解析
本研究以“CDK1在PDAC干细胞中的功能及调控机制”为核心科学问题,采用“数据库分析→细胞功能验证→蛋白质组学筛选→分子机制解析→体内外联合治疗验证”的闭环技术路线,明确CDK1通过STAT3维持PDAC干细胞特性的分子机制,解析非瑟酮靶向CDK1的翻译后修饰调控作用,验证联合治疗的协同效果。
3.1 CDK1在胰腺癌干细胞中的功能验证
本环节核心目标是明确CDK1与PDAC患者预后的相关性,以及其对PDAC干细胞特性的调控作用。研究首先通过GEPIA数据库分析PDAC组织与癌旁组织中CDK1的表达差异,以及CDK1表达与患者总生存期、无病生存期的相关性;随后构建吉西他滨耐药PANC-1细胞系,通过KEGG和GO富集分析差异表达基因,明确CDK1在耐药细胞中的表达特征;采用siRNA沉默PDAC细胞系(PANC-1、HPC-Y5)中的CDK1,通过蛋白质免疫印迹(Western Blot)检测干细胞标志物CD44、Sox2的表达,流式细胞术检测CD44+/CD24+干细胞亚群比例,球形成实验检测干细胞成球能力;同时构建CDK1稳定敲低的PANC-1细胞系,建立裸鼠异种移植模型,观察肿瘤生长情况。
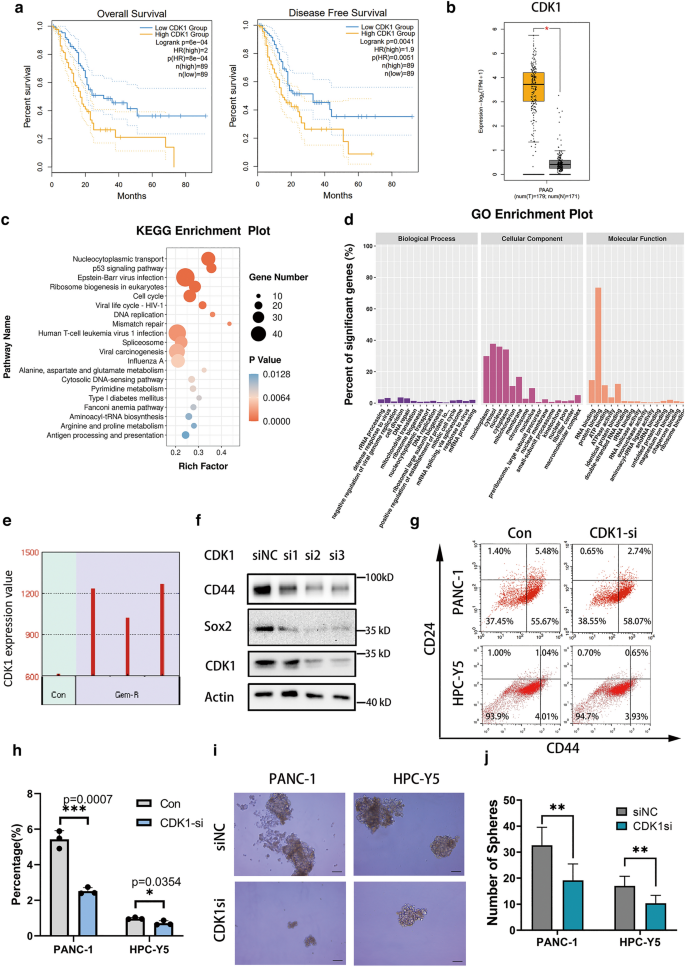
实验结果显示,PDAC组织中CDK1表达显著高于癌旁组织(P<0.05),高表达CDK1的PDAC患者总生存期和无病生存期显著缩短(n=89,P<0.05);吉西他滨耐药PANC-1细胞中CDK1 mRNA表达显著上调;沉默CDK1后,CD44、Sox2的蛋白表达水平显著降低,CD44+/CD24+细胞比例较对照组减少(n=3,P<0.05),球形成数量和大小显著降低(n=6,P<0.01);裸鼠异种移植模型中,CDK1敲低组的肿瘤重量显著低于对照组(n=5,P<0.05)。实验所用关键产品:Abcam的CDK1抗体(货号ab133327)、CD44抗体(货号ab50137)、Sox2抗体(货号ab92494),BD Biosciences的FITC-CD44(货号555478)、PE-CD24(货号555428),Essen BioScience的IncuCyte Live-Cell成像系统等。
3.2 非瑟酮对胰腺癌干细胞特性的抑制作用验证
本环节核心目标是验证非瑟酮对PDAC干细胞特性的抑制效果,筛选其调控的关键信号通路。研究首先采用AutoDock Vina进行分子对接模拟,分析非瑟酮与CDK1的结合模式,通过分子动力学(MD)模拟验证结合稳定性;随后用100μM非瑟酮处理PANC-1和HPC-Y5细胞48小时,通过RNA测序分析干细胞相关基因的表达变化,采用蛋白质免疫印迹验证CD44、Sox2、Oct4、Nanog等干细胞标志物的表达,球形成实验检测干细胞成球能力,流式细胞术检测CD44+/CD24+细胞比例;同时采用稳定同位素标记细胞培养(SILAC)定量蛋白质组学分析非瑟酮处理后差异表达的蛋白,通过磷酸化蛋白芯片检测差异磷酸化蛋白的变化。
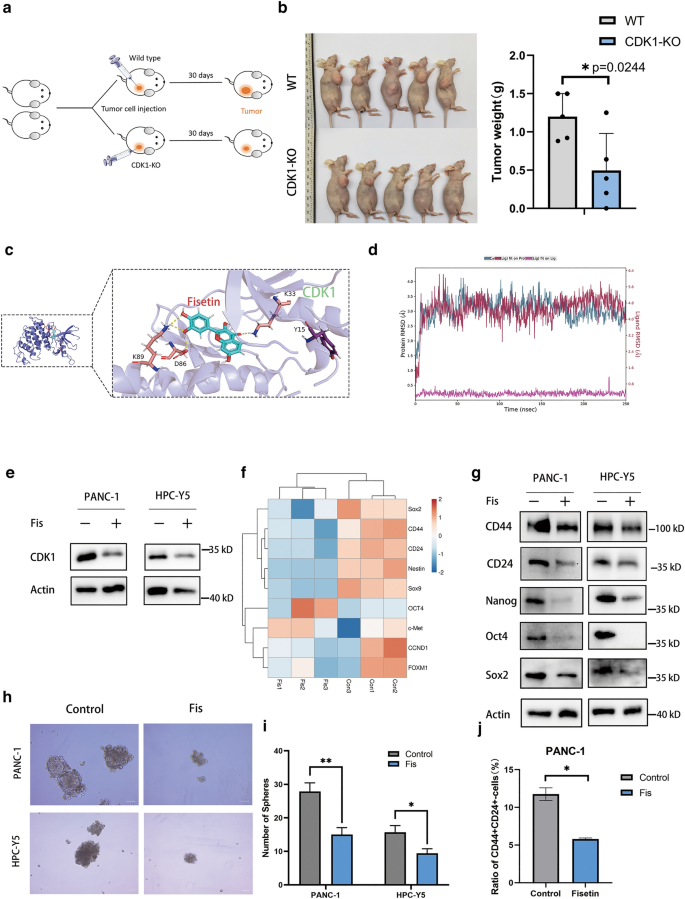
实验结果显示,非瑟酮可结合CDK1的激酶口袋结构域,结合能为-8.5 kcal/mol,MD模拟显示结合模式稳定;非瑟酮处理后,干细胞相关基因SOX2、NANOG、OCT4等的表达显著下调,CD44、Sox2、Oct4、Nanog的蛋白表达水平降低,CD44+/CD24+细胞比例较对照组减少(n=3,P<0.05),成球数量和大小显著降低(n=3,P<0.05);蛋白质组学分析显示非瑟酮抑制蛋白激酶相关信号通路,磷酸化芯片检测到CDK1 Y15和STAT3 Y705位点的磷酸化水平显著降低。实验所用关键产品:Selleck的非瑟酮(货号S2298),Full Moon BioSystems的Phospho Explorer Antibody Array,Thermo的SILAC Protein Quantitation Kit等。
3.3 CDK1的翻译后修饰调控机制解析
本环节核心目标是明确非瑟酮对CDK1翻译后修饰的调控作用,以及乙酰化对CDK1激酶活性的影响。研究采用赖氨酸乙酰化蛋白质组学分析非瑟酮处理后CDK1的乙酰化位点变化;构建CDK1 K33R(去乙酰化突变体,赖氨酸突变为精氨酸)和K33Q(乙酰化模拟突变体,赖氨酸突变为谷氨酰胺)质粒,通过免疫共沉淀(Co-IP)验证CDK1与组蛋白去乙酰化酶3(HDAC3)的相互作用,采用原位邻近连接分析(PLA)检测蛋白相互作用的亚细胞定位;通过蛋白质免疫印迹检测CDK1乙酰化对STAT3磷酸化水平的影响。
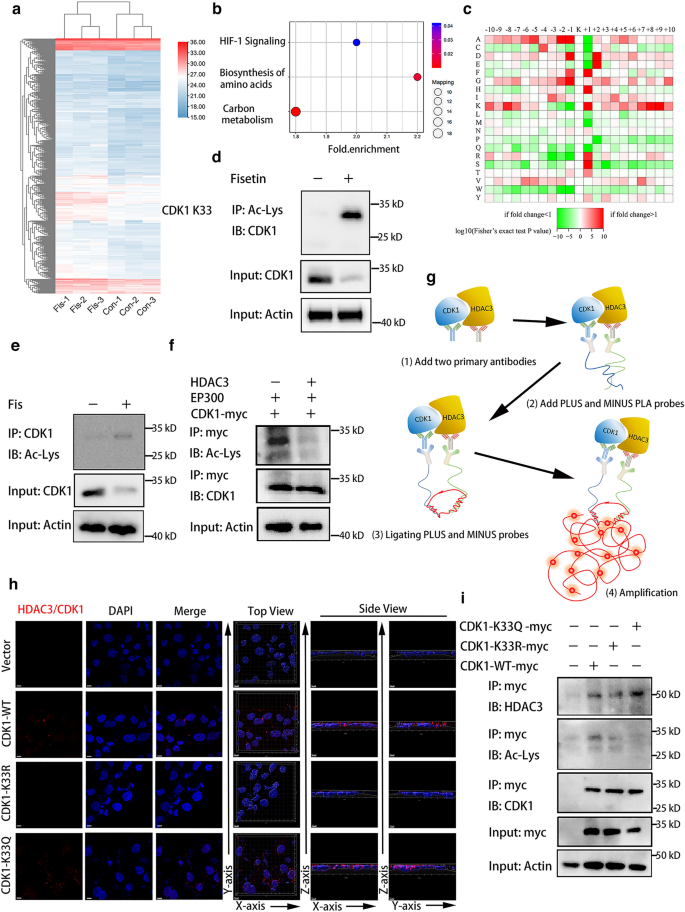
实验结果显示,非瑟酮处理后CDK1 K33位点的乙酰化水平显著上调1.2倍以上(P<0.05);HDAC3可与CDK1直接结合,且HDAC3过表达可显著降低CDK1 K33位点的乙酰化水平;K33Q突变体与HDAC3的结合能力显著增强,K33R突变体与HDAC3的结合能力显著减弱;CDK1 K33乙酰化或突变可显著抑制STAT3 Y705位点的磷酸化水平,降低干细胞标志物CD44、Sox2的表达。实验所用关键产品:PTM Bio的乙酰化赖氨酸抗体(货号PTM-105),Sigma-Aldrich的Duolink PLA试剂盒,Abcam的HDAC3抗体(货号ab32369)等。
3.4 CDK1-STAT3通路调控胰腺癌干细胞的机制验证
本环节核心目标是明确CDK1通过STAT3调控PDAC干细胞特性的分子机制。研究采用siRNA沉默PDAC细胞中的STAT3,或过表达STAT3野生型质粒,通过蛋白质免疫印迹检测干细胞标志物CD44、Sox2的表达,球形成实验检测干细胞成球能力;通过PLA和Co-IP验证CDK1与STAT3的相互作用,检测CDK1 K33突变体对STAT3磷酸化的影响;同时沉默或过表达HDAC3,检测其对CDK1乙酰化及STAT3磷酸化的调控作用。
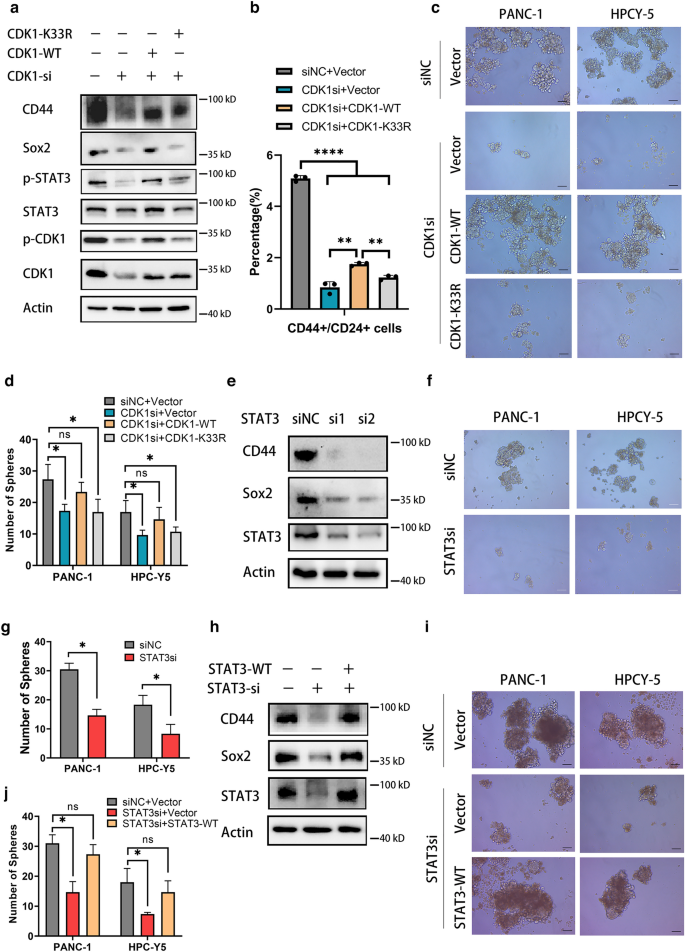
实验结果显示,沉默STAT3可显著降低CD44、Sox2的蛋白表达水平,抑制PDAC细胞的成球能力(n=3,P<0.05),过表达STAT3野生型可逆转CDK1敲低对干细胞特性的抑制效果;CDK1与STAT3在细胞核和细胞质中均存在相互作用,K33R或K33Q突变体可显著减弱这种相互作用,并抑制STAT3 Y705位点的磷酸化;HDAC3沉默可降低CDK1 Y15和STAT3 Y705的磷酸化水平,抑制PDAC细胞的成球能力(n=4,P<0.05),过表达HDAC3则显著增强干细胞特性。实验所用关键产品:Abcam的STAT3抗体(货号ab109085)、磷酸化STAT3抗体(货号ab76315),Ribobio的siRNA转染试剂等。
3.5 非瑟酮联合吉西他滨的体内外抗肿瘤效果验证
本环节核心目标是验证非瑟酮联合吉西他滨对PDAC的协同抗肿瘤作用,明确其机制与干细胞抑制的相关性。研究采用CompuSyn软件计算非瑟酮与吉西他滨的联合指数(CI),通过IncuCyte实时细胞分析系统检测细胞增殖情况;球形成实验检测联合处理对PDAC干细胞成球能力的影响,流式细胞术检测CD44+/CD24+细胞比例;构建KPC(KrasLSL-G12D; p53LSL-R172H; Pdx-1-Cre)小鼠原位移植瘤模型,给予非瑟酮(160mg/kg)、吉西他滨(50mg/kg)单独或联合处理,每两天给药一次,共处理21天,观察肿瘤生长情况,通过免疫组化检测肿瘤组织中CDK1、p-STAT3、CD44、Sox2的表达。
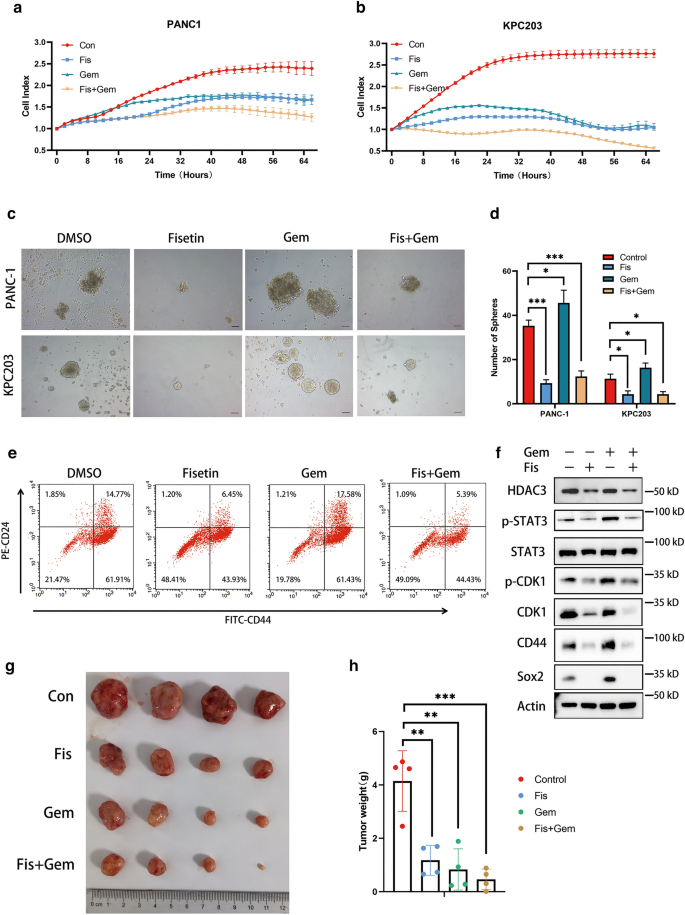
实验结果显示,100μM非瑟酮联合10μM吉西他滨的联合指数CI<1,表明具有协同抗肿瘤作用;联合处理可显著抑制PANC-1和KPC203细胞的增殖,逆转吉西他滨诱导的CD44+/CD24+细胞比例升高,降低干细胞标志物的表达;KPC小鼠模型中,联合治疗组的肿瘤体积和重量显著低于单独治疗组,联合组肿瘤重量为0.466±0.384g,对照组为4.145±1.134g(n=4,P<0.001);免疫组化显示联合治疗组肿瘤组织中CDK1、p-STAT3、CD44、Sox2的表达显著下调。实验所用关键产品:Essen BioScience的IncuCyte Live-Cell成像系统,Olympus的CX31显微镜,免疫组化相关试剂盒等。
4. Biomarker研究及发现成果
本研究涉及的Biomarker包括PDAC干细胞表面标志物CD44+/CD24+亚群、功能调控分子CDK1及其K33乙酰化形式、STAT3及其Y705磷酸化形式,通过“数据库筛选→细胞功能验证→蛋白质组学鉴定→体内外验证”的完整逻辑链条,明确其在PDAC干细胞调控及治疗中的价值。
CD44+/CD24+亚群来源于PDAC细胞系和临床样本,通过流式细胞术进行定量检测,可反映PDAC细胞的干细胞特性;CDK1的表达通过GEPIA数据库分析、蛋白质免疫印迹、免疫组化进行检测,其K33乙酰化水平通过乙酰化蛋白质组学、免疫共沉淀进行检测;STAT3 Y705磷酸化水平通过磷酸化芯片、蛋白质免疫印迹、免疫组化进行检测。特异性方面,CDK1高表达与PDAC患者不良预后显著相关(GEPIA数据库n=89,P<0.05),CD44+/CD24+亚群比例在吉西他滨耐药细胞中显著升高,非瑟酮处理后该比例降低(n=3,P<0.05);敏感性方面,非瑟酮处理后CDK1 K33乙酰化水平上调1.2倍以上(P<0.05),STAT3 Y705磷酸化水平显著降低。
本研究的核心成果在于,首次揭示CDK1 K33位点乙酰化是调控STAT3通路的关键翻译后修饰事件,CDK1通过激活STAT3维持PDAC干细胞的自我更新能力;非瑟酮可通过诱导CDK1 K33乙酰化和泛素化降解,阻断CDK1-STAT3信号通路,显著抑制PDAC干细胞特性;非瑟酮与吉西他滨联合可协同逆转吉西他滨诱导的干细胞耐药,体内外实验均显示联合治疗可显著抑制肿瘤生长。该成果的创新性在于,首次将CDK1的乙酰化修饰与PDAC干细胞调控关联,明确非瑟酮靶向CDK1的分子机制,为PDAC的联合治疗提供了新的生物标志物和治疗靶点,具有重要的临床转化潜力。