1. 领域背景与文献引入
文献英文标题:Identification of novel fusion genes in lung cancer using breakpoint assembly of transcriptome sequencing data;发表期刊:Genome Biology;影响因子:未公开;研究领域:肿瘤基因组学(肺癌融合基因研究)
基因组重排产生的融合基因是肿瘤发生发展的关键驱动因素,这类事件可通过失活抑癌基因或激活原癌基因促进肿瘤进展。领域共识:1986年BCR-ABL融合基因的发现开启了肿瘤靶向治疗时代,伊马替尼的应用成为慢性粒细胞白血病治疗的里程碑;2010年克唑替尼获批用于EML4-ALK阳性非小细胞肺癌,进一步证实融合基因作为精准治疗靶点的临床价值。当前研究热点集中于利用转录组测序(RNA-seq)技术系统挖掘肿瘤中的融合基因,但现有检测方法面临诸多挑战:癌症转录组的复杂性、基因表达的高动态范围及测序误差导致融合基因检测的假阳性率高;多数计算工具仅依赖读对分析或拆分读段映射单一策略,灵敏度不足;部分工具计算资源消耗大,且无法兼容常规RNA-seq分析流程,限制了其在大规模样本中的应用。针对这些未解决的核心问题,本研究开发了TRUP(肿瘤样本适配的RNA-seq统一流程),旨在实现融合基因的高灵敏度、高特异性检测,同时平衡计算效率,为肿瘤融合基因的系统鉴定提供新工具。
2. 文献综述解析
本文综述部分围绕肿瘤融合基因的临床价值、RNA-seq检测融合基因的技术现状及局限性展开,以“现有工具的技术策略-性能短板-临床应用限制”为分类维度,系统梳理领域研究进展。
现有研究表明,配对末端转录组测序(PE RNA-seq)是鉴定融合转录本的有力工具,已成功发现多个具有临床价值的融合基因,如EML4-ALK、ROS1融合等。现有检测方法主要分为三类:基于读对分析的方法,通过比对读对的映射距离或方向异常判断融合事件,优势是计算速度较快,局限性是无法精确定位断点;基于拆分读段映射的方法,通过读段跨断点的部分映射识别融合,优势是断点定位精准,局限性是短读段易产生大量假阳性候选;结合两种策略的方法,一定程度上提升了灵敏度,但仍存在假阳性率高、计算资源消耗大的问题。部分工具如BreakFusion虽引入了从头组装策略提升准确性,但存在计算限制,无法应用于大规模样本。此外,多数工具的定制化映射结果无法复用,导致常规RNA-seq分析需重复计算,增加了研究成本。
本研究的创新价值在于,首次将拆分读段映射、读对分析与候选区域从头组装三种策略深度整合,开发出兼具高灵敏度、高特异性与计算效率的TRUP流程;同时,TRUP的映射结果可直接用于常规RNA-seq分析,解决了现有工具无法兼容常规转录组分析的痛点,为肿瘤样本的多组学整合分析提供了便利。
3. 研究思路总结与详细解析
本研究的核心目标是开发并验证一款高性能的肿瘤融合基因检测计算工具,核心科学问题是如何在复杂的癌症转录组中精准、高效地鉴定融合转录本,技术路线遵循“工具开发-性能验证-临床样本应用-功能验证”的闭环逻辑:首先整合多种计算策略开发TRUP流程并优化版本,随后通过已知融合基因的细胞系、公开数据集及临床样本验证其性能,与现有工具进行多维度对比,最后将TRUP应用于肺癌样本发现新型融合基因,并通过功能实验验证其生物学意义。
3.1 TRUP计算流程开发与优化
实验目的:开发高灵敏度、高特异性的融合基因检测流程,解决现有工具的性能短板。
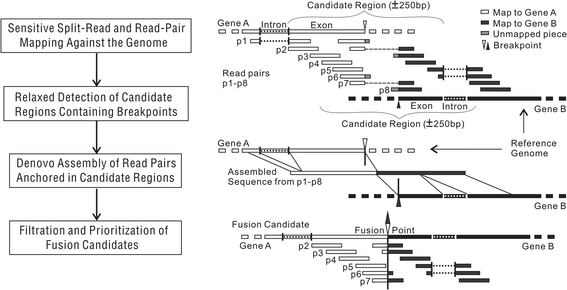
方法细节:首先开发TRUP v1.0版本,采用TopHat进行读段映射,将所有异常读段整合后进行从头组装;基于H3122、H2228等已知EML4-ALK融合的肺癌细胞系验证结果,优化得到TRUP v2.0版本:采用GSNAP或STAR进行拆分读段和读对映射,从映射结果中识别嵌合读段、部分映射读段及异常读对,以宽松标准筛选包含潜在断点的候选区域(±250bp),对每个候选区域的异常读段采用Velvet和Oases进行从头组装,将组装序列通过BLAT比对回参考基因组,基于比对得分、支持读段数等指标过滤并排序融合候选,最终保留高置信度融合事件。
结果解读:TRUP v2.0可检测到TRUP v1.0未发现的28个高置信度融合候选,在7个EML4-ALK阳性样本中,20个候选中有17个经反转录PCR(RT-PCR)、Sanger测序或荧光原位杂交(FISH)验证(验证率85%),表明优化后的流程灵敏度与特异性显著提升。
产品关联:实验所用关键工具:TRUP(开源,https://github.com/ruping/TRUP)、GSNAP、STAR、Velvet、Oases、BLAT;文献未提及具体实验试剂品牌的常规实验环节,领域常规使用Illumina TruSeq建库试剂盒、Trizol等RNA提取试剂。
3.2 TRUP性能验证与工具对比
实验目的:验证TRUP的灵敏度、特异性及计算效率,并与现有主流融合检测工具进行对比。
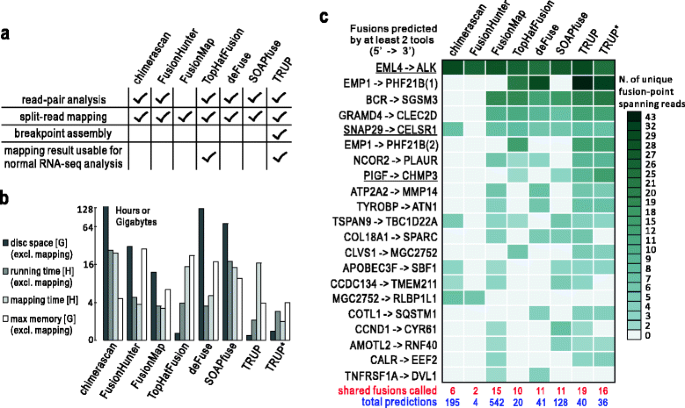
方法细节:将TRUP v2.0应用于公开的小细胞肺癌RNA-seq数据集,检测新型融合基因;选取肺癌样本S00054,与chimerascan、FusionHunter等7款主流工具(排除2款因计算限制无法运行的工具)进行对比,从计算资源消耗(磁盘空间、内存、运行时间)、检测灵敏度、结果可复用性等维度评估性能;采用“至少两款工具共同检测到的融合”作为金标准,评估各工具的召回率与精确率。
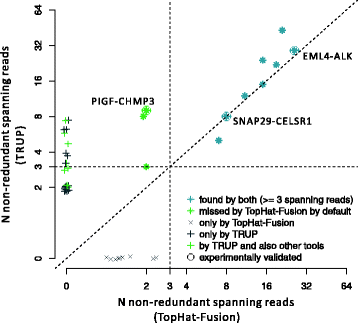
结果解读:TRUP在小细胞肺癌细胞系N417和H187中分别检测到CREBBP和TAF6L的失活融合,与领域内组蛋白修饰基因重排参与肿瘤发生的结论一致;工具对比显示,TRUP的计算资源消耗显著低于其他工具,即使采用GSNAP映射,其整体运行效率仍更优,且映射结果可直接用于常规RNA-seq分析(仅TRUP与TopHat-Fusion支持结果复用);以金标准评估,TRUP的召回率与精确率的调和均值为0.62,显著高于TopHat-Fusion的0.49,且能检测到其他工具遗漏的验证阳性融合(如PIGF-CHMP3),表明TRUP在灵敏度与特异性间实现了更优平衡。
产品关联:文献未提及此环节的具体实验产品,领域常规使用Linux服务器进行计算分析,采用Illumina HiSeq系列平台产生的RNA-seq数据。
3.3 EML4-ALK阳性样本次级融合基因检测
实验目的:探究EML4-ALK阳性肺癌样本中是否存在伴随的次级融合基因,解析其基因组起源。
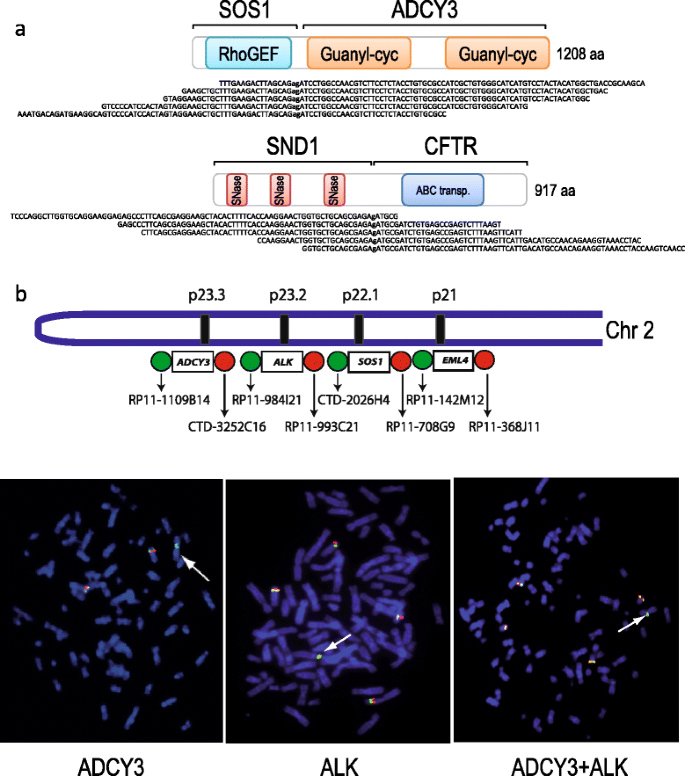
方法细节:采用TRUP分析H3122、H2228细胞系及5例EML4-ALK阳性肺癌临床样本的RNA-seq数据,筛选伴随的融合基因;通过断点分离荧光原位杂交验证融合事件的基因组水平改变,针对H3122细胞系中的SOS1-ADCY3融合,同时进行ALK与ADCY3的断点分离荧光原位杂交实验,验证其是否由同一基因组事件产生。
结果解读:在H3122中检测到SOS1-ADCY3融合,H2228中检测到SND1-CFTR和DCBLD2-STXBP5L融合,4例临床样本中均检测到至少1个次级框内融合基因;断点分离荧光原位杂交实验显示,H3122细胞系中ALK与ADCY3的重排信号重叠,表明SOS1-ADCY3与EML4-ALK由同一染色体2的基因组事件产生;拷贝数数据分析显示,多数融合事件存在明确的基因组断点,排除反式剪接的可能,提示肺癌中的融合事件多为平衡易位。
产品关联:实验所用关键产品:ALK、ADCY3等基因的BAC克隆荧光原位杂交探针,Lipofectamine RNAiMax(Life Technologies),RASSF8小鼠单克隆抗体(Santa Cruz, 4B1),HRP标记山羊抗小鼠二抗(Millipore),β-actin HRP标记抗体(Santa Cruz);BCA蛋白定量试剂盒(Pierce),蛋白酶抑制剂(Roche),磷酸酶抑制剂(Merck),PMSF(Carl Roth)。
3.4 RASSF8融合基因功能验证
实验目的:验证TRUP发现的RASSF8融合基因的生物学功能,探究其在肿瘤发生中的作用。
方法细节:采用TRUP分析17例肺腺癌样本的RNA-seq数据,筛选RASSF8融合事件;在表达野生型RASSF8的H1395肺癌细胞系中,通过esiRNA沉默RASSF8表达,设置EGFP转染组为对照,培养6天后计数细胞数量,通过蛋白质免疫印迹验证RASSF8的沉默效率;同时分析骨肉瘤细胞系KPD的RNA-seq数据,检测RASSF8融合事件。
结果解读:在1例EGFR/KRAS野生型的吸烟患者肺腺癌样本中检测到ASUN-RASSF8失活融合,在KPD骨肉瘤细胞系中检测到RASSF8-MARS失活融合;RASSF8沉默后,H1395细胞增殖率较对照组增加60%以上(P<0.0001,n=3),蛋白质免疫印迹显示RASSF8未完全沉默,提示低水平的RASSF8表达即可促进细胞增殖,支持RASSF8作为抑癌基因的功能。
产品关联:实验所用关键产品:esiRNA,Lipofectamine RNAiMax(Life Technologies),RASSF8小鼠单克隆抗体(Santa Cruz, 4B1),HRP标记山羊抗小鼠二抗(Millipore),β-actin HRP标记抗体(Santa Cruz);BCA蛋白定量试剂盒(Pierce),蛋白酶抑制剂(Roche),磷酸酶抑制剂(Merck),PMSF(Carl Roth)。
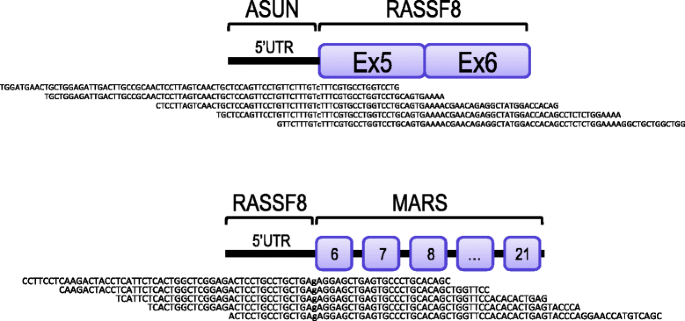
4. Biomarker研究及发现成果解析
本研究中涉及的Biomarker为肿瘤融合基因,包括EML4-ALK阳性样本中的次级融合基因(如SOS1-ADCY3、SND1-CFTR等)及RASSF8失活融合基因,筛选与验证逻辑为“TRUP RNA-seq数据分析→RT-PCR/Sanger测序验证→荧光原位杂交基因组水平验证→功能实验验证”。
Biomarker定位:次级融合基因属于伴随驱动融合的基因组事件,RASSF8失活融合属于抑癌基因失活事件;筛选流程为:通过TRUP从肺癌RNA-seq数据中识别融合候选,经支持读段数、比对得分等过滤后得到高置信度候选,再通过RT-PCR、Sanger测序验证转录本水平的融合,通过荧光原位杂交验证基因组水平的重排,最后通过功能实验验证其生物学意义。
研究过程详述:次级融合基因来源于EML4-ALK阳性肺癌细胞系及临床样本,RASSF8融合基因来源于肺腺癌临床样本及骨肉瘤细胞系;验证方法包括:RT-PCR与Sanger测序验证融合转录本的存在,荧光原位杂交验证基因组重排,蛋白质免疫印迹验证基因表达水平,细胞增殖实验验证功能;特异性与敏感性方面,TRUP在已知融合样本中的验证率达85%,在工具对比中对金标准融合的召回率高于其他工具;RASSF8沉默后细胞增殖率增加60%以上(P<0.0001,n=3),显示其功能显著性。
核心成果提炼:首次在EML4-ALK阳性肺癌样本中发现伴随的次级框内融合基因,证实部分次级融合与EML4-ALK由同一基因组事件产生,提示肺癌基因组重排的复杂性;首次在肺癌和骨肉瘤中发现RASSF8的失活重排,通过功能实验证实RASSF8具有抑癌功能,其失活可促进肿瘤细胞增殖(细胞增殖率增加60%以上,P<0.0001,n=3);TRUP工具可作为肿瘤融合基因检测的高效工具,为新型治疗靶点的发现提供技术支撑。