1. 领域背景与文献引入
文献英文标题:SARS-CoV-2 NSP13 helicase suppresses interferon signaling by perturbing JAK1 phosphorylation of STAT1;发表期刊:Cell Bioscience;影响因子:未公开;研究领域:冠状病毒免疫逃逸机制
新型冠状病毒(SARS-CoV-2)引发的新型冠状病毒肺炎(COVID-19)疫情给全球公共卫生体系带来沉重负担,重症患者常因细胞因子释放综合征(CRS)进展为急性呼吸窘迫综合征(ARDS),而JAK-STAT信号通路在细胞因子产生和干扰素(IFN)抗病毒免疫应答中发挥核心调控作用。目前JAK抑制剂被用于缓解重症患者的细胞因子风暴,但由于JAK通路同时调控IFN诱导的抗病毒基因(ISG)表达,这类治疗存在抑制免疫应答与增强病毒感染的权衡问题。领域共识:SARS-CoV-2通过编码多种病毒蛋白实现免疫逃逸,已有研究证实NSP1、NSP6、ORF6等多个蛋白可通过不同机制抑制IFN信号通路,但NSP13解旋酶的具体作用机制仍存在争议,其对I型和II型IFN信号的广谱抑制缺乏统一的分子机制解释,因此亟需深入解析NSP13调控JAK-STAT通路的分子细节,为优化JAK抑制剂治疗和开发新型抗病毒药物提供理论依据。
2. 文献综述解析
作者以SARS-CoV-2病毒蛋白的免疫逃逸机制为核心,按蛋白功能靶点分类梳理了现有研究成果,重点对比了不同病毒蛋白调控IFN信号通路的差异,并指出NSP13的作用机制尚未明确的研究空白。
现有研究系统筛选并鉴定了SARS-CoV-2中可拮抗IFN信号的关键蛋白,包括NSP1、NSP6、NSP13、NSP14、ORF3a、ORF6等,这些蛋白通过不同节点调控JAK-STAT通路:NSP1通过关闭宿主翻译机器或下调TYK2和STAT2表达抑制ISG产生,NSP6直接阻碍STAT1磷酸化,NSP14诱导IFNAR1的溶酶体降解,ORF6通过劫持核孔蛋白Nup98抑制STAT1核转位。这些研究的优势在于全面覆盖了病毒蛋白的免疫逃逸功能,为理解SARS-CoV-2的致病机制提供了多个关键靶点,但局限性在于部分蛋白的具体分子机制解析不够深入,尤其是NSP13,此前研究仅提出其可能通过抑制STAT1磷酸化、下调IFNAR1表达或与STAT1结合发挥作用,但未明确其调控JAK1对STAT1磷酸化的具体方式,也未解释其对I型和II型IFN信号的广谱抑制的统一机制。本文通过对比现有研究的未解决问题,明确了核心创新点:首次揭示NSP13通过与STAT1直接结合,不影响JAK1-STAT1复合物形成,但特异性抑制JAK1对STAT1的磷酸化,且该功能依赖其解旋酶活性,完善了NSP13的免疫逃逸机制模型,为其广谱抑制IFN信号提供了统一的分子解释。
3. 研究思路总结与详细解析
本文的研究目标是明确SARS-CoV-2 NSP13抑制IFN信号通路的分子机制,核心科学问题是NSP13如何调控JAK-STAT通路中STAT1磷酸化的关键步骤,技术路线遵循“功能验证→机制定位→分子互作→活性依赖”的闭环逻辑,通过细胞实验、分子生物学实验和突变体分析逐步解析其作用机制。
3.1 IFN信号抑制功能验证
本环节的实验目的是验证NSP13对I型和II型IFN信号通路的广谱抑制作用。研究人员首先在转染效率较高的HEK293T细胞中,将不同剂量的NSP13表达质粒与pISRE-Luc(响应I型IFN)或pGAS-Luc(响应II型IFN)报告基因质粒共转染,用IFN-β或IFN-γ刺激后检测荧光素酶活性;同时在肺腺癌A549细胞(SARS-CoV-2易感细胞系)中过表达NSP13,用IFN-β或IFN-γ刺激后,通过实时荧光定量聚合酶链反应(RT-qPCR)检测ISG15、OAS1、CXCL10、CXCL11、IRF1等ISG的mRNA表达水平。结果显示,NSP13以剂量依赖方式显著抑制IFN-β诱导的ISRE启动子活性,以及IFN-γ诱导的ISRE和GAS启动子活性(n=3,P<0.05或P<0.01或P<0.001);RT-qPCR结果显示,NSP13可显著下调IFN-β诱导的ISG15、OAS1表达,以及IFN-γ诱导的CXCL11、IRF1表达,同时抑制两种IFN诱导的CXCL10表达(n=3,P<0.05或P<0.01或P<0.001),证实NSP13对I型和II型IFN信号均具有抑制作用。
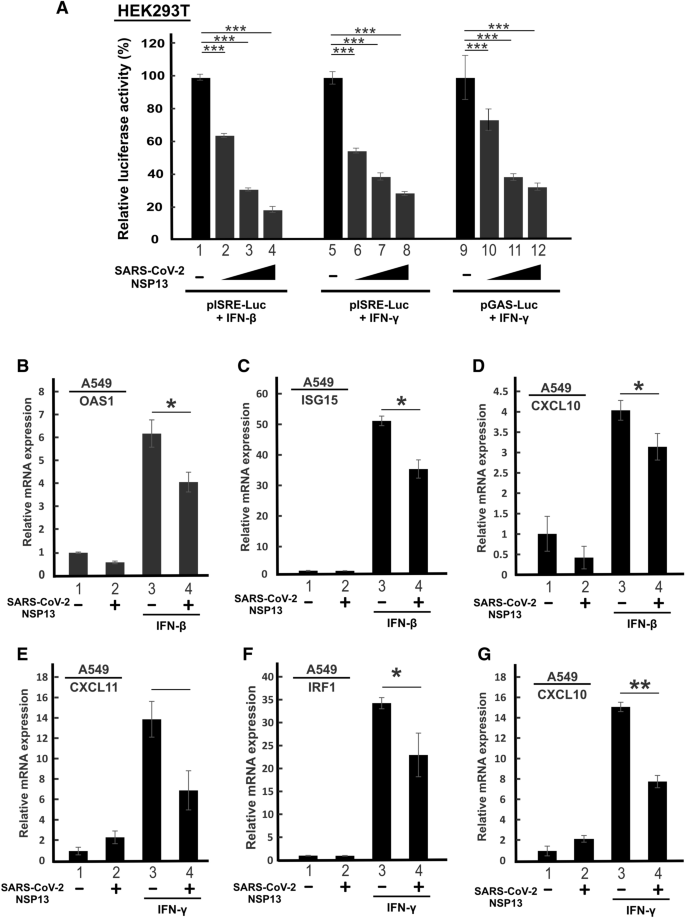
产品关联:实验所用关键试剂包括Genejuice转染试剂(MilliporeSigma)、Lipofectamine 3000转染试剂(Thermo Fisher Scientific)、IFN-β1a和IFN-γ(PBL Assay Science)、TB Green Premix Ex Taq(TaKaRa)等。
3.2 STAT核转位与磷酸化水平检测
本环节的实验目的是定位NSP13在JAK-STAT通路中的具体作用节点。研究人员在A549细胞中转染带有V5标签的NSP13,用IFN-β或IFN-γ刺激30分钟后,通过免疫荧光染色观察内源性STAT1和STAT2的亚细胞定位;同时在HEK293T细胞中转染NSP13,用IFN-β或IFN-γ刺激10、20、30、45分钟后,通过蛋白质印迹(Western blot)检测JAK1、STAT1、STAT2的磷酸化水平和总蛋白表达量。免疫荧光结果显示,IFN-β刺激可诱导STAT1和STAT2向细胞核转位,IFN-γ刺激仅诱导STAT1核转位,而过表达NSP13可完全阻止上述核转位过程;蛋白质印迹结果显示,NSP13对IFN-β或IFN-γ诱导的JAK1磷酸化无明显影响,但显著抑制STAT1的磷酸化水平,对STAT2的磷酸化无显著作用,说明NSP13的作用靶点是STAT1的磷酸化步骤,而非JAK1的激活。
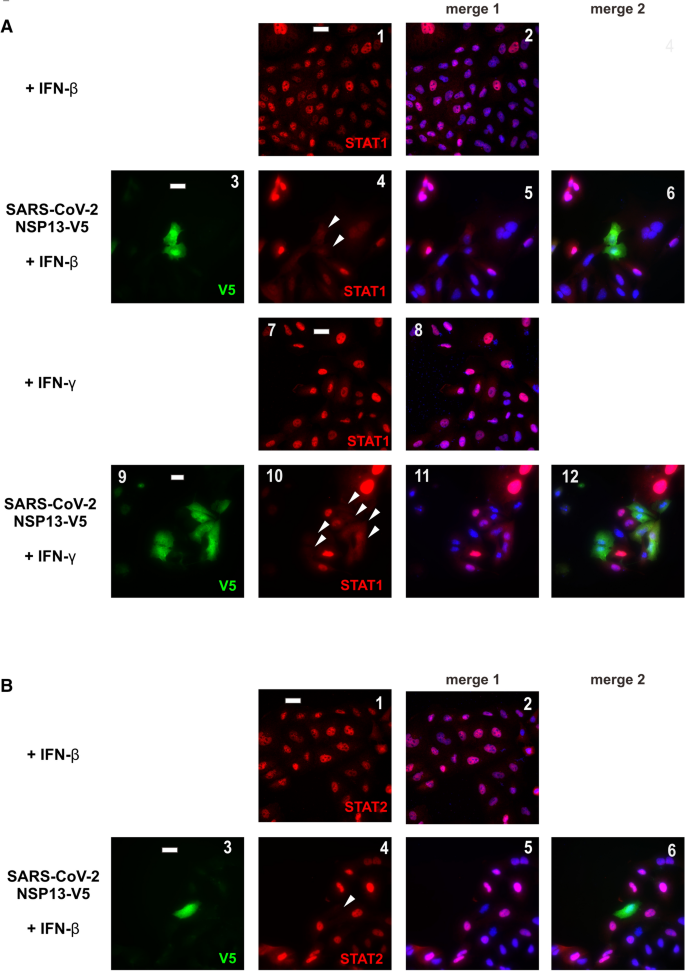
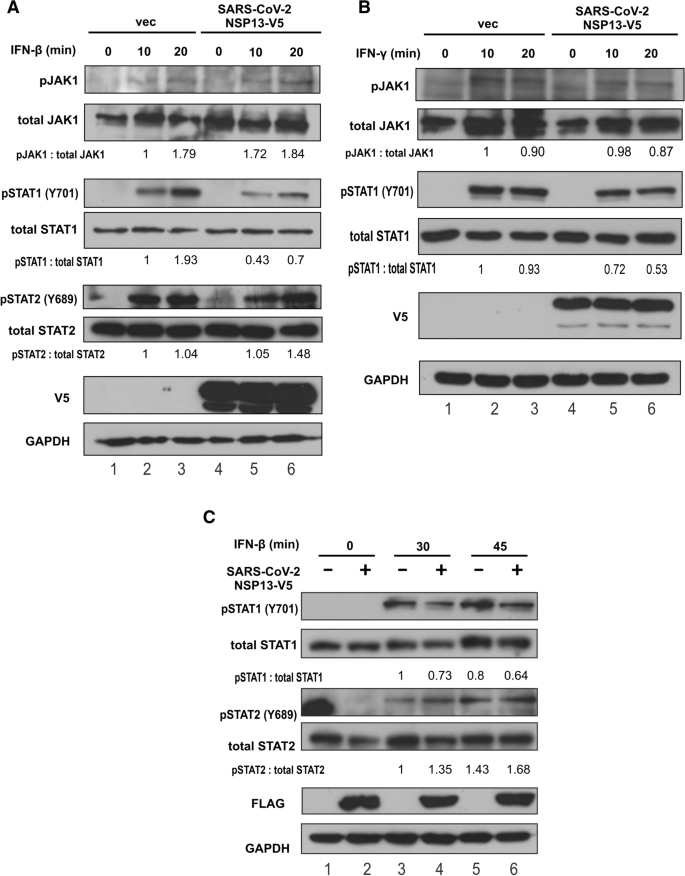
产品关联:使用的关键抗体包括抗STAT1、STAT2、JAK1(Santa Cruz Biotechnology),抗磷酸化STAT1、JAK1(Cell Signaling Technology),抗磷酸化STAT2(R&D Systems),抗V5标签抗体(Thermo Fisher Scientific)等。
3.3 NSP13与STAT1的分子互作分析
本环节的实验目的是解析NSP13抑制STAT1磷酸化的分子机制。研究人员在HEK293T细胞中共转染NSP13与JAK1或STAT1,通过免疫共沉淀(Co-IP)检测蛋白间的相互作用;同时进行体外激酶实验,将免疫沉淀得到的NSP13与重组JAK1、STAT1蛋白及ATP共孵育,通过蛋白质印迹检测STAT1的磷酸化水平。免疫共沉淀结果显示,NSP13可与STAT1直接结合,但不与JAK1结合,且NSP13的表达不影响JAK1与STAT1的复合物形成;体外激酶实验结果显示,NSP13可显著抑制JAK1对STAT1的磷酸化,说明NSP13通过与STAT1结合,直接干扰JAK1对STAT1的磷酸化过程,而非通过阻断JAK1与STAT1的结合发挥作用。
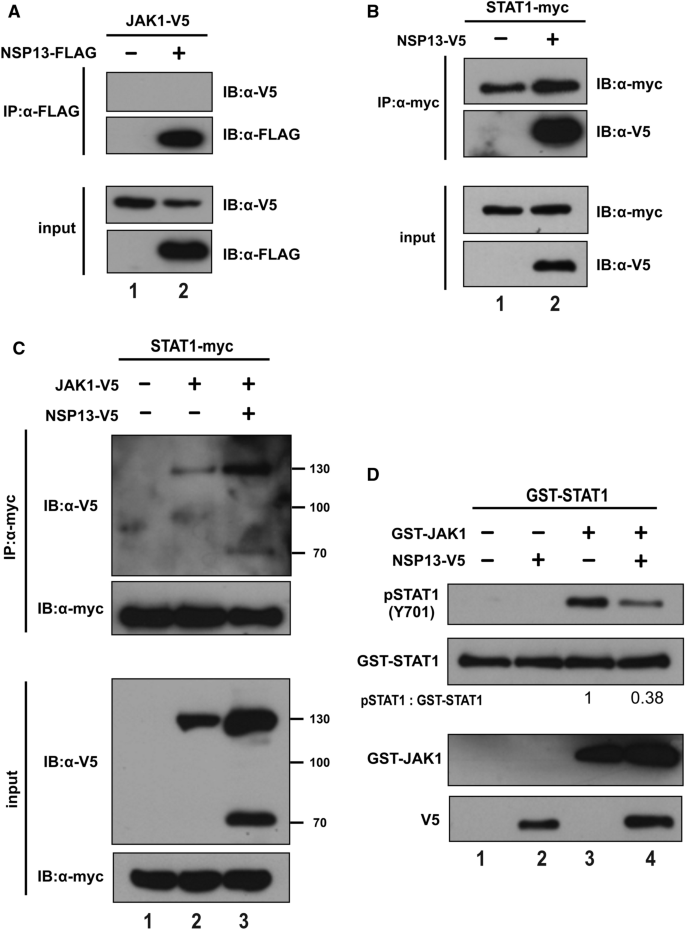
产品关联:实验所用关键试剂包括Q5®定点突变试剂盒(New England Biolabs)、重组STAT1(Sino Biological)、重组JAK1(MilliporeSigma)等。
3.4 解旋酶活性对IFN抑制的必要性验证
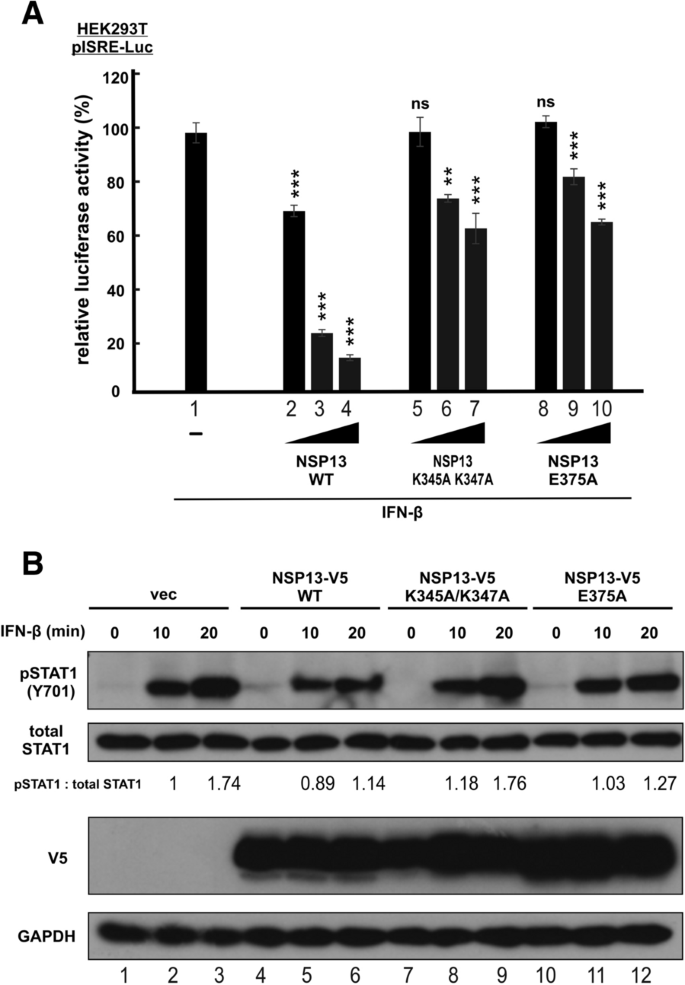
本环节的实验目的是确认NSP13的解旋酶活性在IFN信号抑制中的作用。研究人员构建了NSP13的两个功能缺陷突变体:核酸结合缺陷突变体K345A K347A和NTP酶(解旋酶核心活性)缺陷突变体E375A,在HEK293T细胞中表达这些突变体,通过报告基因实验检测其对IFN-β诱导的ISRE启动子活性的影响,同时通过蛋白质印迹检测其对STAT1磷酸化的抑制作用。报告基因实验显示,与野生型NSP13相比,两个突变体均显著丧失对ISRE启动子活性的抑制能力(n=3,P<0.01或P<0.001);蛋白质印迹结果显示,K345A K347A突变体完全丧失对STAT1磷酸化的抑制作用,E375A突变体仅保留部分抑制能力,说明NSP13的解旋酶活性是其抑制IFN信号通路的必要条件,其核酸结合和NTP酶活性均参与调控STAT1的磷酸化。
产品关联:使用Q5®定点突变试剂盒(New England Biolabs)构建突变体,其余试剂与前述实验一致。
4. Biomarker研究及发现成果
本文未涉及疾病诊断、预后或疗效预测相关的传统生物标志物研究,聚焦于病毒蛋白NSP13作为免疫逃逸关键分子的功能机制解析,其核心发现成果为明确了NSP13是SARS-CoV-2广谱抑制I型和II型IFN信号的核心分子,其作用机制为通过与STAT1直接结合,不影响JAK1-STAT1复合物形成,但特异性抑制JAK1对STAT1的磷酸化,且该功能依赖其解旋酶活性(核酸结合和NTP酶活性)。
该成果的创新性在于首次揭示了NSP13调控JAK-STAT通路的具体分子细节,解释了其对I型和II型IFN信号的广谱抑制的统一机制,为开发靶向NSP13的抗病毒药物提供了新的靶点和理论依据。由于本文为细胞和分子水平的机制研究,未涉及临床样本的检测分析,因此无生物标志物的特异性、敏感性数据,也无预后相关的统计学结果(如风险比HR、ROC曲线AUC值等)。