1. 领域背景与文献引入
文献英文标题:Disruption of zinc transporter ZnT3 transcriptional activity and synaptic vesicular zinc in the brain of Huntington’s disease transgenic mouse;发表期刊:Cell Bioscience;影响因子:未公开;研究领域:神经退行性疾病(亨廷顿病)的突触功能障碍与锌稳态调控。
领域共识:亨廷顿病(HD)是一种常染色体显性遗传神经退行性疾病,1993年首次发现其致病机制为HD基因第一外显子的CAG三核苷酸重复序列异常扩增(>36次),编码突变亨廷顿蛋白(mHtt)。当前HD研究热点聚焦于早期突触功能障碍的发生机制、可用于早期诊断的生物标志物开发,以及靶向突触稳态的新型治疗策略。领域内未解决的核心问题包括:mHtt如何在神经元死亡前诱导突触功能障碍,脑内锌稳态失衡在HD早期认知障碍中的具体调控机制,以及锌转运蛋白在其中的作用尚未明确。
针对上述研究空白,本研究旨在揭示mHtt对脑内突触囊泡锌稳态的调控作用及分子机制,其学术价值在于首次阐明HD中ZnT3转录活性受抑制的机制,为HD早期突触功能障碍提供新的病理解释,同时为开发靶向锌稳态的HD治疗策略提供理论依据。
2. 文献综述解析
作者以“HD发病机制的核心环节→突触功能障碍的早发性特征→锌稳态与突触功能的紧密关联→HD中锌稳态研究的缺失”为逻辑主线,系统梳理了领域内现有研究的进展与不足。
现有研究支持HD的核心致病机制为mHtt的毒性作用,包括蛋白聚集、转录调控异常及突触功能损伤,其中突触功能障碍早于神经元死亡数年,是HD早期症状(如认知障碍、精神症状)的主要原因。技术方法上,转基因小鼠模型和细胞模型被广泛用于HD发病机制研究,结合分子生物学、影像学技术可精准解析突触功能变化,但现有研究存在局限性:仅发现HD患者血液锌水平升高,未明确脑内锌稳态的变化及调控机制,且缺乏对锌转运蛋白ZnT3转录调控的深入研究,无法解释HD早期突触锌丢失的原因。
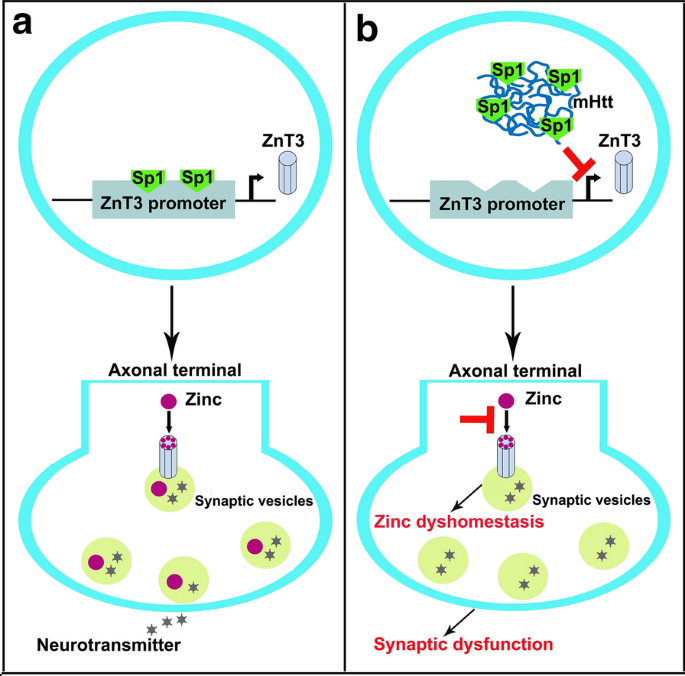
本研究的创新价值在于,通过对比现有研究中HD锌稳态的研究空白,首次揭示mHtt通过抑制转录因子Sp1与ZnT3启动子的结合,下调ZnT3表达,导致突触囊泡锌丢失,进而引发早期突触功能障碍。这一发现填补了HD中锌稳态调控机制的空白,为HD的早期干预提供了新的靶点。
3. 研究思路总结与详细解析
本研究的整体研究框架为“提出假设→体内外实验验证锌及ZnT3的变化→分子机制解析Sp1对ZnT3的转录调控→验证mHtt对Sp1结合的抑制作用→总结机制与功能关联”,围绕“mHtt如何调控突触锌稳态”这一核心科学问题,通过多层面实验形成完整的逻辑闭环。
3.1 转基因小鼠脑内锌水平检测
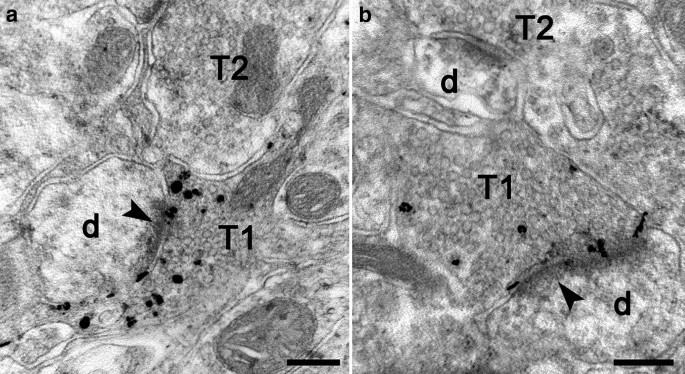
本环节的实验目的是验证HD转基因小鼠脑内总锌和突触囊泡锌的变化,明确锌稳态是否失衡。实验方法为:选取20周龄N171-82Q HD转基因小鼠及同龄野生型小鼠,采用火焰原子吸收光谱法(FAAS)检测脑皮层、纹状体、海马的总锌含量,同时采用自动金属显影法(AMG)结合光镜、电镜观察突触囊泡锌的分布与含量。实验结果显示,转基因小鼠三个脑区的总锌水平显著降低(n=3,P<0.05);光镜下AMG染色显示,野生型小鼠海马CA1-CA3区、皮层、纹状体的锌染色强度高,而转基因小鼠对应区域染色极弱;电镜下可见野生型小鼠纹状体突触囊泡内存在大量锌颗粒,转基因小鼠突触囊泡内锌颗粒数量显著减少。产品关联:文献未提及具体实验产品,领域常规使用原子吸收光谱仪、透射电子显微镜等仪器。
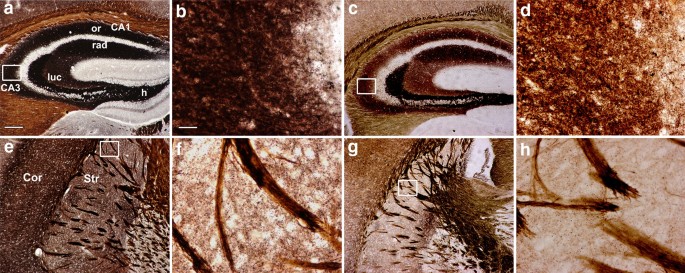
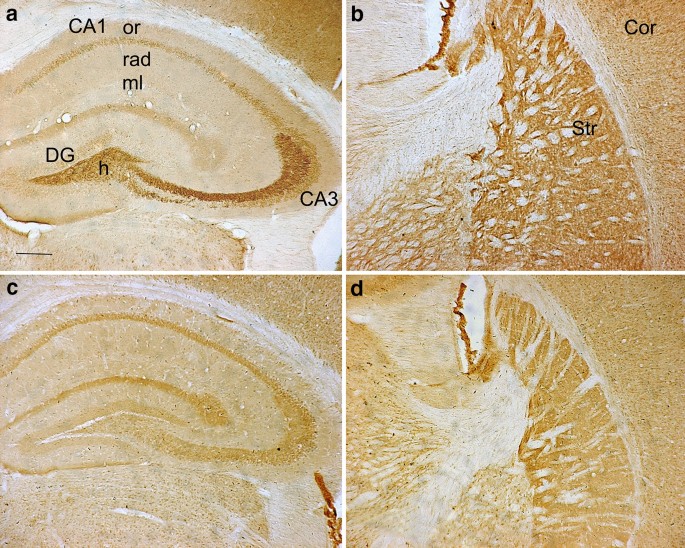
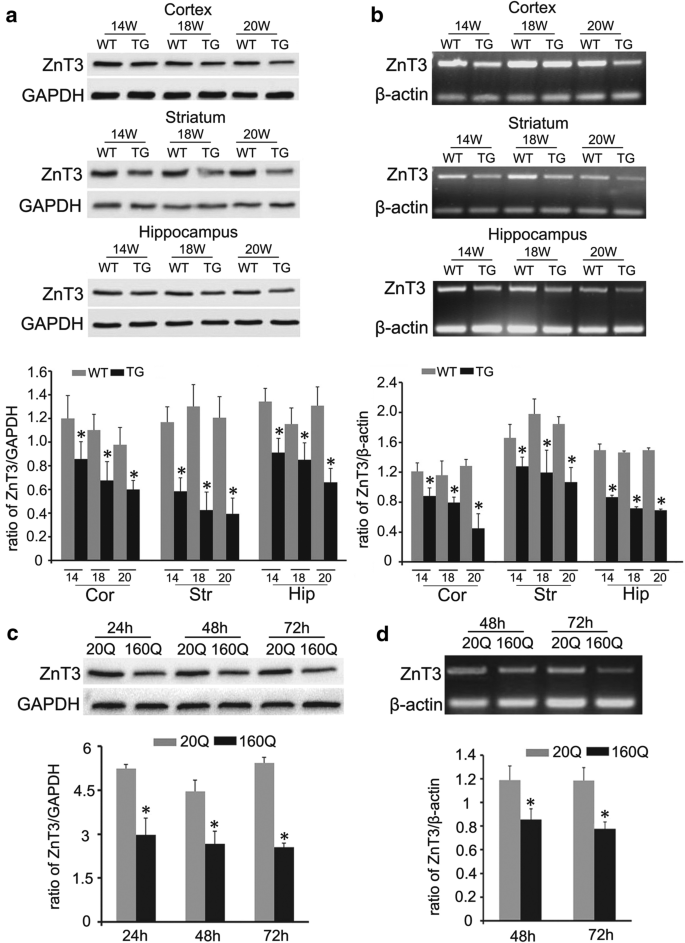
3.2 ZnT3表达水平的体内外验证
本环节的实验目的是明确突触囊泡锌减少是否由ZnT3表达下调导致,因为ZnT3是负责将锌转运至突触囊泡的关键蛋白。实验方法为:采用免疫组化检测20周龄转基因小鼠脑内ZnT3的分布与表达,通过蛋白质免疫印迹(Western blot)和逆转录聚合酶链反应(RT-PCR)检测14、18、20周龄转基因小鼠三个脑区的ZnT3蛋白和mRNA水平,同时检测表达mHtt的BHK细胞(160Q细胞)中的ZnT3表达变化。实验结果显示,野生型小鼠脑内三个脑区的ZnT3免疫反应性强,而转基因小鼠对应区域免疫反应性极弱;14-20周龄转基因小鼠的ZnT3蛋白和mRNA水平均显著低于同龄野生型小鼠(n=4,P<0.05);160Q细胞中的ZnT3蛋白和mRNA水平也显著低于表达正常亨廷顿蛋白的20Q细胞(n=4,P<0.05)。产品关联:实验所用关键产品:Proteintech的ZnT3多克隆抗体(1:100)、Sigma-Aldrich的RIPA裂解液、Pierce Thermo Scientific的增强化学发光(ECL)试剂盒。
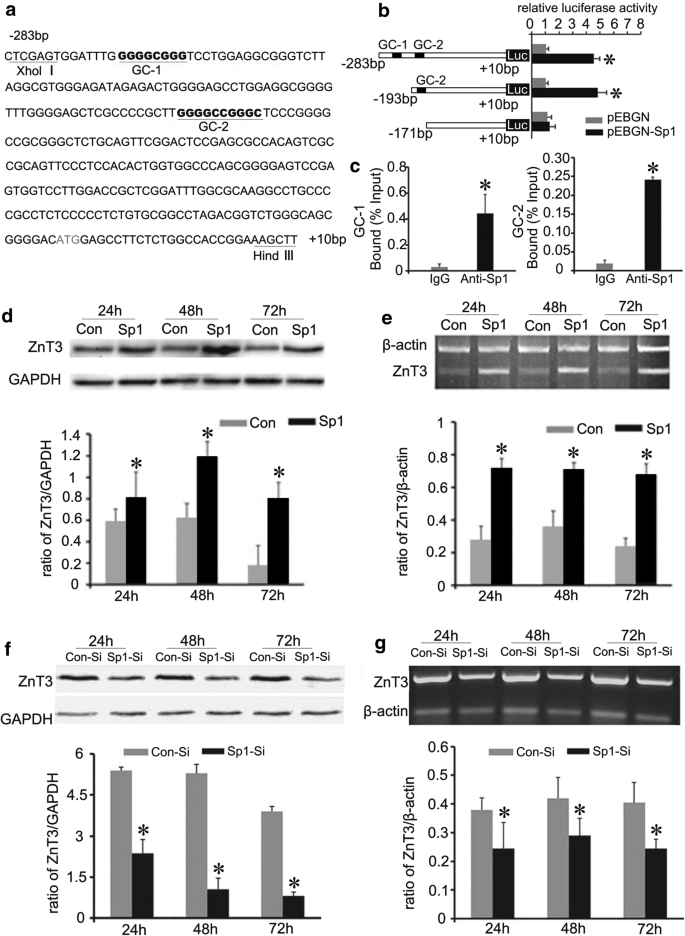
3.3 Sp1对ZnT3的转录调控验证
本环节的实验目的是确定ZnT3的转录调控因子及具体机制,解析ZnT3表达下调的转录层面原因。实验方法为:通过生物信息学分析ZnT3启动子区域的转录因子结合位点,发现两个GC盒(Sp1结合位点);采用双荧光素酶报告基因实验验证Sp1对ZnT3启动子的激活作用,通过染色质免疫沉淀(ChIP)实验验证Sp1与ZnT3启动子的直接结合;同时通过过表达和敲低Sp1,检测ZnT3表达的变化。实验结果显示,双荧光素酶实验表明Sp1可显著激活含GC盒的ZnT3启动子活性(n=3,P<0.05),而不含GC盒的启动子活性无变化;ChIP实验显示Sp1与ZnT3启动子的两个GC盒区域结合显著高于IgG对照(n=3,P<0.05);过表达Sp1可显著上调BHK细胞中ZnT3的蛋白和mRNA水平(n=4,P<0.05),敲低Sp1则显著下调ZnT3表达(n=4,P<0.05)。产品关联:实验所用关键产品:Promega的双荧光素酶报告基因检测系统、Millipore的Magna ChIP A Kit、Abcam的Sp1 ChIP级抗体。
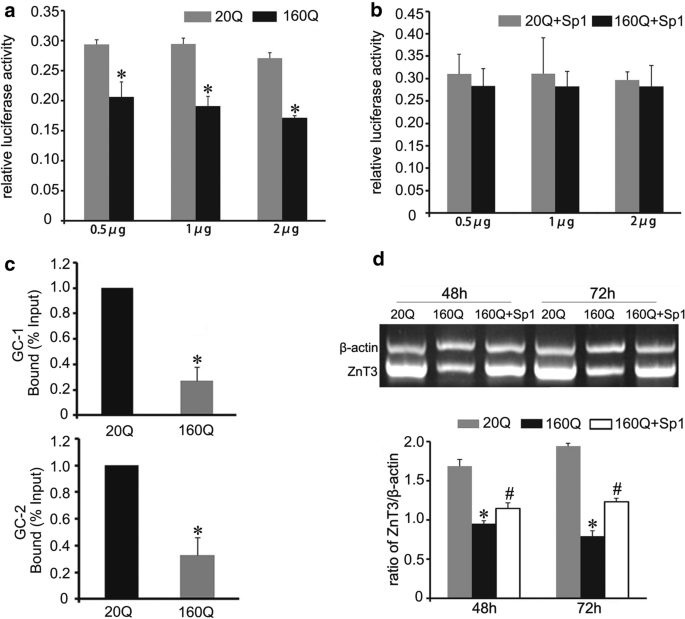
3.4 mHtt对Sp1-ZnT3调控轴的影响验证
本环节的实验目的是验证mHtt是否通过抑制Sp1与ZnT3启动子的结合,下调ZnT3表达。实验方法为:采用双荧光素酶报告基因实验,将不同浓度的mHtt载体与ZnT3启动子报告质粒共转染BHK细胞,检测启动子活性;通过ChIP实验检测160Q细胞中Sp1与ZnT3启动子的结合情况;同时过表达Sp1,检测是否能逆转mHtt对ZnT3表达的抑制作用。实验结果显示,mHtt以剂量依赖方式抑制ZnT3启动子活性(n=3,P<0.05);160Q细胞中Sp1与ZnT3启动子的结合显著低于20Q细胞(n=3,P<0.05);过表达Sp1可显著逆转160Q细胞中ZnT3的mRNA水平下调(n=3,P<0.05)。产品关联:文献未提及具体实验产品,领域常规使用Lipofectamine 2000转染试剂、实时荧光定量PCR仪等。
4. Biomarker研究及发现成果
本研究涉及的生物标志物包括锌转运蛋白ZnT3(蛋白型生物标志物)和突触囊泡锌(功能型生物标志物),通过体内外实验系统验证了其在HD早期的变化及调控机制,为HD的早期诊断和治疗提供了潜在靶点。
Biomarker定位:ZnT3作为HD早期突触功能障碍的潜在生物标志物,其筛选与验证逻辑为“转基因小鼠模型验证→细胞模型验证→分子机制解析→功能关联确认”;突触囊泡锌作为功能型生物标志物,反映了突触功能的损伤程度。研究过程详述:Biomarker来源为N171-82Q HD转基因小鼠的脑皮层、纹状体、海马组织及表达mHtt的160Q BHK细胞;验证方法包括免疫组化、蛋白质免疫印迹、逆转录聚合酶链反应、自动金属显影法;特异性与敏感性数据显示,ZnT3表达在14周龄的转基因小鼠脑内即显著降低(n=4,P<0.05),具有早期检测的潜力,突触囊泡锌在20周龄时三个脑区均显著丢失(n=3,P<0.05)。
核心成果提炼:ZnT3的表达下调与mHtt抑制Sp1结合直接相关,是HD早期突触锌丢失的关键原因;突触囊泡锌丢失可导致谷氨酸介导的兴奋性毒性增强,进而引发突触功能障碍和认知障碍;本研究未提供该Biomarker的临床样本验证数据、风险比(HR)等临床相关数据,需进一步临床研究确认其诊断价值。