1. 领域背景与文献引入
文献英文标题:Combination of EZH2 inhibitor and BET inhibitor for treatment of diffuse intrinsic pontine glioma;发表期刊:Cell Bioscience;影响因子:未公开;研究领域:儿童脑肿瘤(弥漫内生型桥脑胶质瘤)、肿瘤表观遗传学治疗
弥漫内生型桥脑胶质瘤(DIPG)是一种发生于脑干的致命性儿童恶性脑肿瘤,是儿童癌症相关死亡的主要原因之一,其5年生存率不足1%,中位总生存期仅约9个月,1年和2年生存率分别约为30%和10%以下。目前放疗是DIPG的标准治疗方案,虽能短暂改善患者症状,但肿瘤复发迅速,化疗尚未显示出明确获益,因此亟需深入解析DIPG的分子机制以开发新型治疗策略。近年研究发现,约80%的DIPG存在组蛋白H3K27M突变,该突变会导致全局H3K27me3水平降低,但部分抑癌基因仍保留H3K27me3修饰并持续沉默,为表观遗传治疗提供了潜在靶点。EZH2作为PRC2复合物的核心催化亚基,以及BET家族蛋白作为表观调控因子,已被证实与DIPG的发生发展密切相关,但单独使用其抑制剂的治疗效果仍有局限,因此本研究旨在探索EZH2抑制剂与BET抑制剂联合治疗DIPG的协同作用及机制。
2. 文献综述解析
作者围绕DIPG的表观遗传调控靶点,将现有研究分为EZH2靶向治疗和BET靶向治疗两个核心方向展开评述。现有研究显示,EZH2负责催化H3K27me3修饰,H3K27M突变后,部分抑癌基因仍依赖EZH2的活性维持沉默,EZH2抑制剂可在细胞和动物模型中抑制DIPG生长,但单独治疗难以完全阻断肿瘤进展;BET家族蛋白(如BRD2、BRD4)可结合H3K27M-K27ac异源核小体,参与DIPG的致病过程,BET抑制剂JQ-1能有效抑制肿瘤细胞增殖,但单独使用的生存获益有限。现有研究的核心局限性在于,尚未探索EZH2与BET抑制剂联合治疗的协同效应,也未明确联合治疗对不同表观调控模式基因的特异性作用。本研究的创新价值在于,首次在细胞和动物模型中验证了EZH2抑制剂(EPZ6438)与BET抑制剂(JQ-1)联合治疗DIPG的显著协同效果,通过特异性调控H3K27M异常沉默的抑癌基因,增强抗肿瘤作用,为DIPG的临床联合治疗提供了新的实验依据。
3. 研究思路总结与详细解析
本研究整体遵循“模型构建→体外功能验证→机制探究→体内疗效验证”的闭环逻辑,核心研究目标是明确EZH2与BET抑制剂联合治疗DIPG的效果及表观调控机制,解决的核心科学问题是联合治疗如何通过表观遗传修饰调控抑癌基因表达以抑制肿瘤生长。
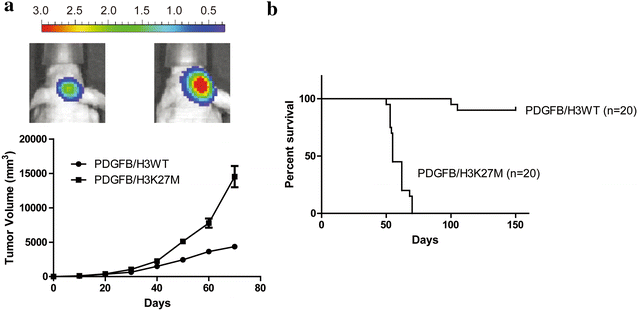
3.1 H3K27M突变DIPG细胞模型构建与验证
本环节的核心目标是构建模拟人类DIPG的细胞模型,验证H3K27M突变的致癌性。实验方法为从E12.5小鼠胚胎背侧前脑分离神经干细胞(NSCs),转染HA标记的PDGFB和Flag标记的H3K27M或H3K27wt,培养后采用免疫印迹(Western blot)检测H3K27me3水平,软琼脂克隆形成实验检测成瘤能力,细胞计数试剂盒-8(CCK-8)检测细胞增殖,随后将细胞原位移植到6周龄雌性BALB/c小鼠桥脑构建动物模型。结果显示,H3K27M组NSCs的全局H3K27me3水平显著降低,软琼脂克隆形成数显著高于H3K27wt组(n=3,P<0.001),细胞增殖速度更快;小鼠原位移植后,H3K27M组肿瘤体积更大,生存率显著低于H3K27wt组(每组n=20),证实H3K27M突变足以诱导DIPG发生。
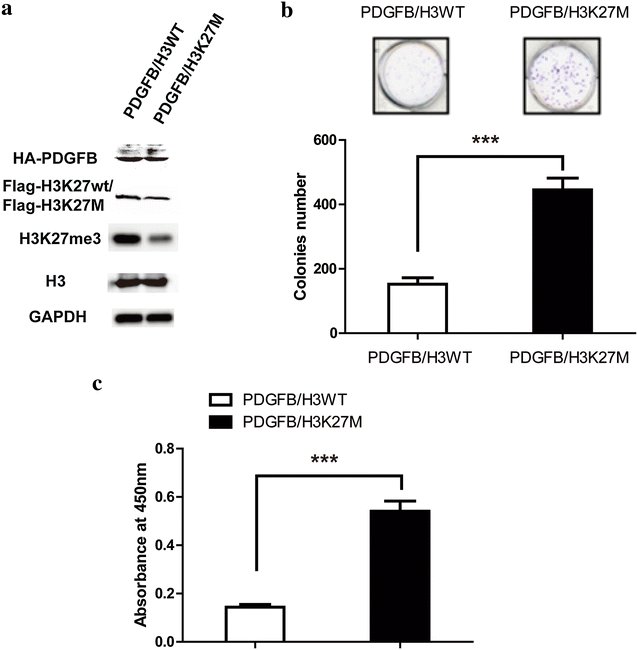
实验所用关键产品:EZH2抑制剂EPZ6438、BET抑制剂JQ-1购自Selleck Chemicals;免疫印迹所用Flag-tag、HA-tag、H3K27me3等抗体购自Sigma-Aldrich;CCK-8试剂盒购自Dojindo Laboratories。
3.2 联合抑制剂对DIPG细胞增殖与凋亡的影响
本环节的核心目标是验证EZH2与BET抑制剂单独及联合使用对肿瘤细胞增殖和凋亡的调控作用。实验方法为用EPZ6438(3μM)、JQ-1(300nM)单独或联合处理PDGFB/H3K27M NSCs,通过细胞计数和CCK-8检测细胞增殖能力,采用Annexin V-FITC/PI染色结合流式细胞术检测细胞凋亡水平。结果显示,单独使用任一抑制剂均能显著降低细胞增殖活性,联合处理后的增殖抑制效果更显著(n=3,P<0.001);单独处理可显著诱导细胞凋亡,联合处理后凋亡率进一步升高(n=3,P<0.001),证实EZH2和BET蛋白的活性对DIPG细胞生长至关重要,联合抑制可产生协同抗肿瘤效应。
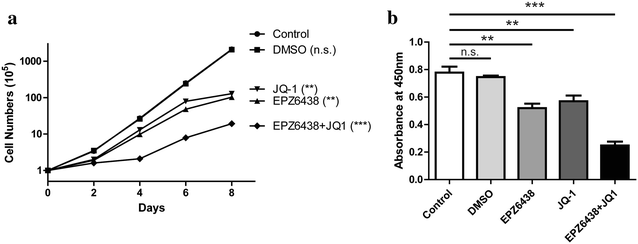
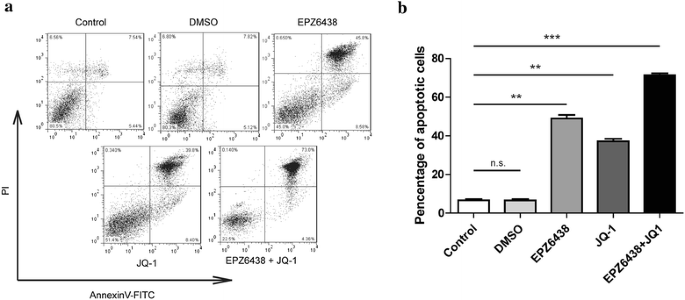
3.3 联合抑制剂的表观调控机制研究
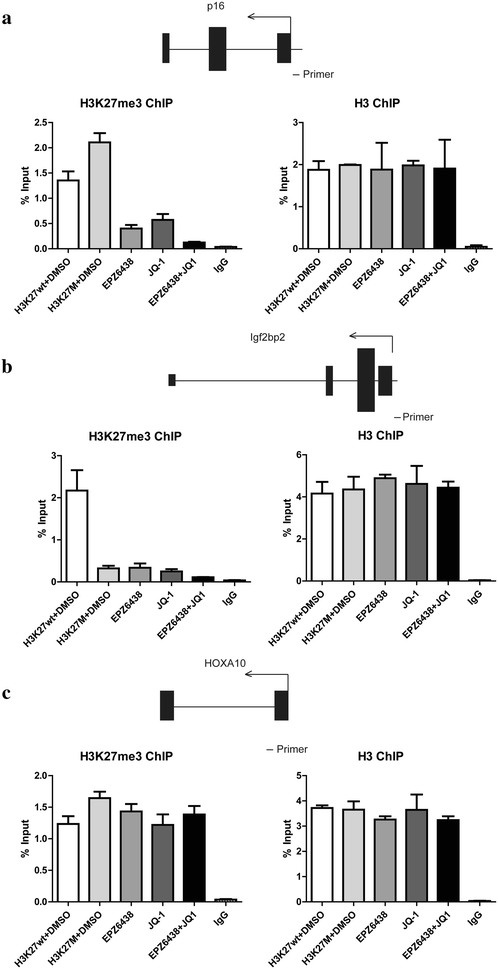
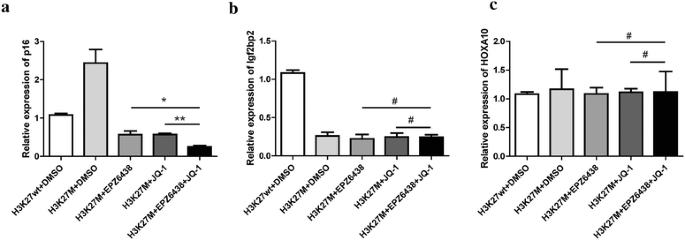
本环节的核心目标是探究联合治疗对不同表观调控模式基因的特异性作用机制。实验方法为采用染色质免疫沉淀-定量PCR(ChIP-qPCR)检测抑癌基因p16^INK4A、Igf2bp2、HOXA10启动子区的H3K27me3水平,通过实时定量PCR(qRT-PCR)检测基因表达水平。结果显示,p16^INK4A启动子区H3K27me3水平在H3K27M细胞中显著升高,单独使用任一抑制剂均能显著降低该水平,联合处理后H3K27me3完全消除,p16^INK4A表达显著上调;Igf2bp2启动子区H3K27me3在H3K27M细胞中已丢失,抑制剂处理无明显影响;HOXA10作为PRC2靶基因,其启动子区H3K27me3水平保留,抑制剂处理无显著变化,证实联合治疗可特异性调控H3K27M异常沉默的抑癌基因。
3.4 动物模型中联合治疗的生存获益验证
本环节的核心目标是在体内验证联合治疗的疗效。实验方法为将PDGFB/H3K27M NSCs原位移植到小鼠桥脑,肿瘤形成5天后,分别腹腔注射EPZ6438(250mg/kg)、JQ-1(50mg/kg)单独或联合治疗,每日监测小鼠生存率。结果显示,单独治疗可延长小鼠生存时间,联合治疗后150天仍有50%的小鼠存活,生存获益显著优于单独治疗组(每组n=20),证实联合治疗在体内具有显著的抗肿瘤效果。

4. Biomarker研究及发现成果
本研究涉及的核心Biomarker包括H3K27M突变及抑癌基因p16^INK4A启动子区的H3K27me3修饰。其中H3K27M是DIPG的核心驱动突变,筛选逻辑基于临床样本中80%的突变频率,本研究通过细胞和动物模型验证了其致癌性;p16^INK4A启动子区的H3K27me3作为治疗响应Biomarker,筛选逻辑为H3K27M突变后该区域异常保留H3K27me3导致基因沉默,是潜在的治疗靶点。研究过程中,H3K27M突变体通过细胞转染获得,采用免疫印迹验证H3K27me3水平变化,ChIP-qPCR验证p16^INK4A启动子区H3K27me3的调控,联合治疗后该区域H3K27me3完全消除,p16^INK4A表达显著上调。核心成果为,H3K27M突变是DIPG的关键致癌驱动因子,联合EZH2和BET抑制剂可特异性调控H3K27M异常沉默的抑癌基因,显著抑制肿瘤生长并延长小鼠生存时间,为DIPG的联合表观遗传治疗提供了新的策略,同时p16^INK4A启动子区的H3K27me3可作为潜在的治疗响应Biomarker,为后续临床研究提供了参考。