1. 领域背景与文献引入
文献英文标题:Overexpression of PTEN suppresses lipopolysaccharide-induced lung fibroblast proliferation, differentiation and collagen secretion through inhibition of the PI3-K-Akt-GSK3beta pathway;发表期刊:Cell Bioscience;影响因子:未公开;研究领域:急性肺损伤/肺纤维化分子机制
急性肺损伤(ALI)是临床常见的危重症,可进展为急性呼吸窘迫综合征(ARDS)并伴随弥漫性肺纤维化,最终导致呼吸衰竭,脂多糖(LPS)作为革兰氏阴性菌内毒素是ALI的重要诱因之一。领域共识:肺成纤维细胞的异常增殖、分化为肌成纤维细胞并过度分泌胶原,是肺纤维化发生发展的核心病理环节,磷脂酰肌醇3-激酶-蛋白激酶B(PI3-K-Akt)通路在这一过程中发挥关键调控作用。现有研究已证实LPS可通过Toll样受体4介导激活PI3-K-Akt通路,促进肺成纤维细胞功能异常,但LPS诱导肺纤维化的具体分子调控机制尚未完全阐明。磷酸酶及张力蛋白同源物(PTEN)作为PI3-K通路的负调控因子,其在LPS诱导肺成纤维细胞功能异常中的作用及具体调控通路仍不明确,这是当前领域的核心研究空白。本文旨在探讨PTEN过表达对LPS诱导的肺成纤维细胞增殖、分化及胶原分泌的影响,并明确其是否通过PI3-K-Akt-糖原合成酶激酶3β(GSK3β)通路发挥调控作用,为LPS诱导的肺纤维化提供潜在治疗靶点。
2. 文献综述解析
本文作者按“肺纤维化病理机制→PI3-K-Akt通路的调控作用→PTEN的功能研究现状”的维度梳理领域内现有研究,系统整合了不同方向的研究结论与局限性。现有研究证实,ALI/ARDS向肺纤维化进展的核心是肺成纤维细胞的异常活化,LPS可直接诱导肺成纤维细胞分泌胶原,且这一过程依赖PI3-K-Akt通路的激活;PTEN作为经典的肿瘤抑制因子,通过去磷酸化活性负调控PI3-K-Akt通路,在多种细胞的增殖、凋亡及分化中发挥关键作用,部分研究显示PTEN可调控肺成纤维细胞向肌成纤维细胞分化,但缺乏其在LPS诱导肺纤维化中的直接功能证据。现有研究的局限性在于,未明确PTEN的磷酸酶活性与表达水平在调控肺成纤维细胞功能中的相对重要性,也未系统解析PTEN对PI3-K-Akt-GSK3β通路的完整调控机制,无法为肺纤维化的靶向治疗提供精准的分子靶点。本文的创新价值在于,首次在LPS诱导的小鼠原代肺成纤维细胞模型中,明确PTEN过表达可通过抑制PI3-K-Akt-GSK3β通路,全面逆转LPS诱导的细胞增殖、分化及胶原分泌,且首次证实PTEN的磷酸酶活性比其表达水平对肺成纤维细胞功能的调控更为关键,填补了领域内关于PTEN在LPS诱导肺纤维化中作用机制的研究空白。
3. 研究思路总结与详细解析
本文的研究目标是明确PTEN在LPS诱导肺成纤维细胞功能异常中的调控作用及分子机制,核心科学问题是PTEN是否通过PI3-K-Akt-GSK3β通路抑制LPS诱导的肺成纤维细胞增殖、分化及胶原分泌,技术路线遵循“构建PTEN过表达细胞模型→验证PTEN表达及活性→检测通路关键分子变化→评估细胞功能改变→抑制剂验证通路特异性”的闭环逻辑,实验设计严谨,结果互为验证。
3.1 PTEN过表达细胞模型构建与PTEN功能验证
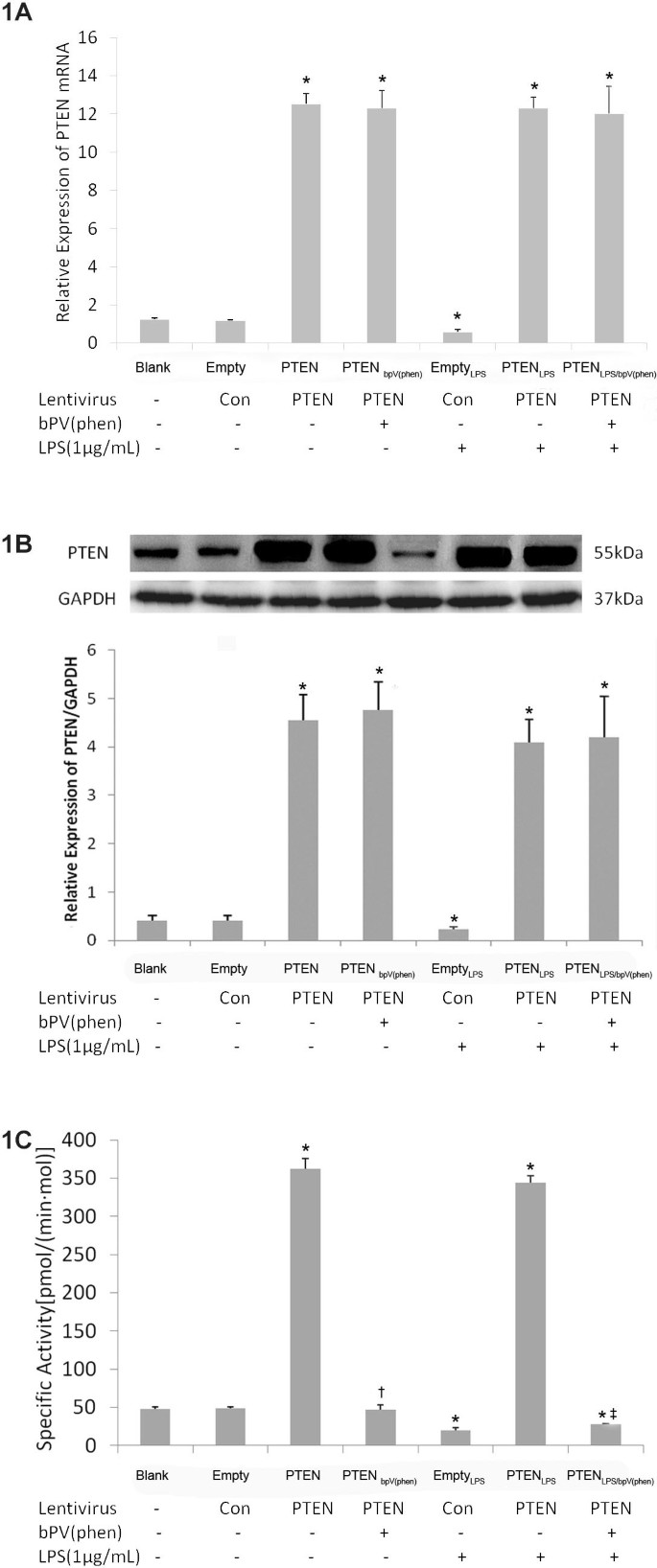
实验目的:构建PTEN过表达的小鼠原代肺成纤维细胞模型,验证LPS处理及PTEN过表达对PTEN表达水平及磷酸酶活性的影响。方法细节:分离培养C57/BL6小鼠原代肺成纤维细胞,将PTEN过表达慢病毒以5×10^4转导单位(TU)/mL的浓度转染细胞48h,随后用1μg/mL LPS处理72h;设置空白对照组、空载体转染组、空载体+LPS组、PTEN过表达组、PTEN过表达+LPS组、PTEN过表达+LPS+PTEN抑制剂bpV(phen)组等;采用实时荧光定量PCR(real-time RT-PCR)检测PTEN mRNA表达,蛋白质免疫印迹(Western blot)检测PTEN蛋白表达,孔雀绿法检测PTEN磷酸酶活性。结果解读:与空载体组相比,空载体+LPS组的PTEN mRNA、蛋白表达及磷酸酶活性显著降低(P<0.05,n=3);与空载体+LPS组相比,PTEN过表达+LPS组的PTEN mRNA、蛋白表达及磷酸酶活性显著升高(P<0.05,n=3);PTEN抑制剂bpV(phen)可显著抑制PTEN过表达+LPS组的磷酸酶活性,但不影响其mRNA及蛋白表达。实验所用关键产品:PTEN过表达慢病毒(GeneChem)、LPS(Sigma)、bpV(phen)(Alexis)、实时荧光定量PCR引物、蛋白质免疫印迹一抗(CST)。
3.2 PI3-K-Akt-GSK3β通路关键分子检测
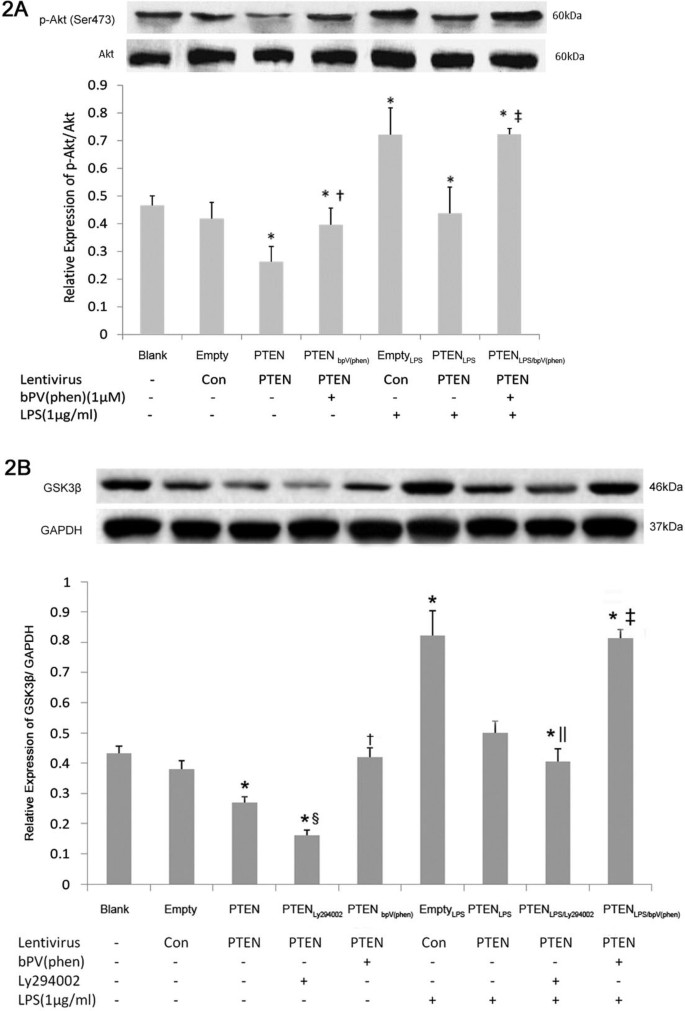
实验目的:验证PTEN过表达对LPS诱导的PI3-K-Akt-GSK3β通路激活的调控作用。方法细节:收集不同处理组细胞的总蛋白,采用蛋白质免疫印迹检测Akt(Ser473)磷酸化水平、GSK3β蛋白表达水平;同时设置PI3-K抑制剂Ly294002处理组(50μmol/L处理1h后加LPS),检测GSK3β表达变化。结果解读:与空载体组相比,空载体+LPS组的Akt磷酸化水平及GSK3β表达显著升高(P<0.05,n=3);与空载体+LPS组相比,PTEN过表达+LPS组的Akt磷酸化水平及GSK3β表达显著降低(P<0.05,n=3),且与未加LPS的组无显著差异;PTEN抑制剂bpV(phen)可逆转PTEN过表达对Akt磷酸化及GSK3β表达的抑制作用,使其恢复至空载体+LPS组水平;PI3-K抑制剂Ly294002可增强PTEN过表达对GSK3β表达的抑制作用(P<0.05,n=3),进一步证实PTEN通过抑制PI3-K-Akt通路下调GSK3β表达。实验所用关键产品:Ly294002(CST)、蛋白质免疫印迹一抗(CST)。
3.3 肺成纤维细胞增殖能力检测
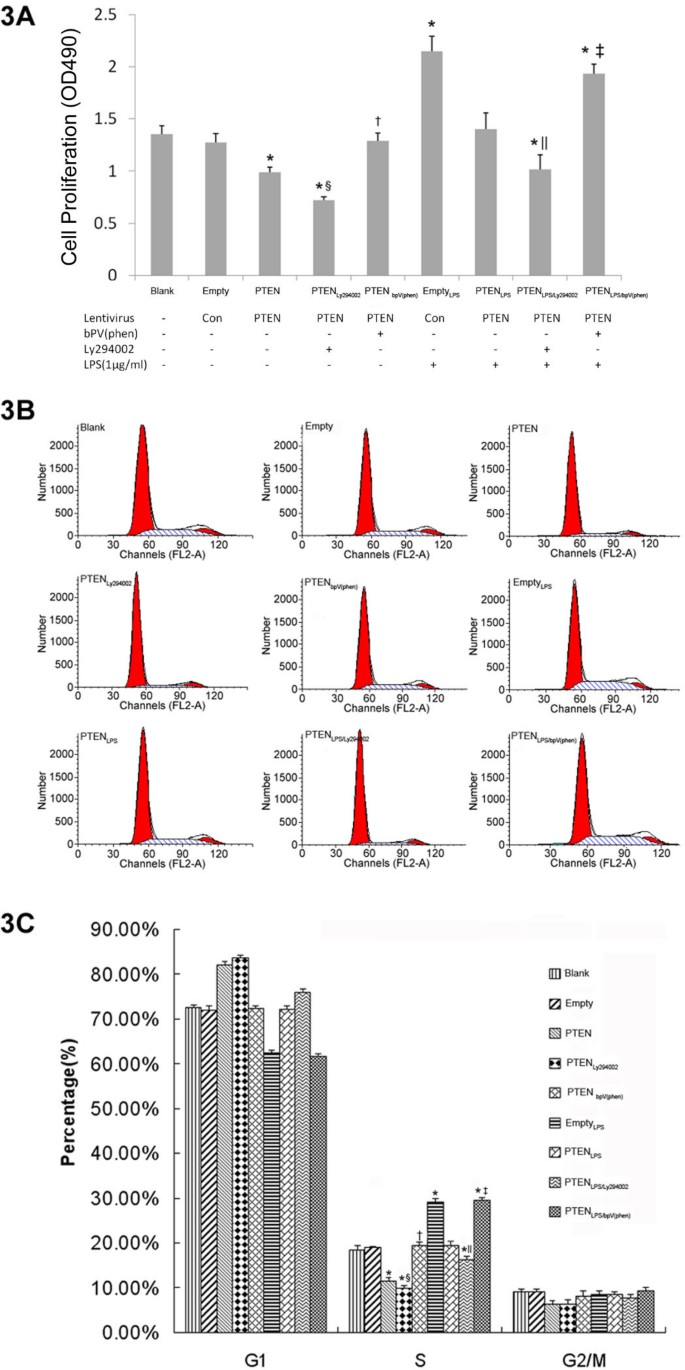
实验目的:评估PTEN过表达对LPS诱导的肺成纤维细胞增殖的影响。方法细节:采用MTT法检测细胞活力,流式细胞术检测细胞周期分布;各组处理同前,MTT实验在LPS处理72h后加入MTT,孵育4h后检测OD490nm值;流式细胞术采用碘化丙啶(PI)染色,分析S期细胞比例。结果解读:与空载体组相比,空载体+LPS组的细胞活力及S期细胞比例显著升高(P<0.05,n=3);与空载体+LPS组相比,PTEN过表达+LPS组的细胞活力及S期细胞比例显著降低(P<0.05,n=3),与未加LPS的组无显著差异;PTEN抑制剂bpV(phen)可逆转PTEN过表达对细胞增殖的抑制作用,PI3-K抑制剂Ly294002可增强PTEN过表达的抑制效果。实验所用关键产品:MTT试剂、PI染色液(Sigma)、流式细胞仪(BD FACSCalibur™)。
3.4 肺成纤维细胞分化及胶原分泌检测
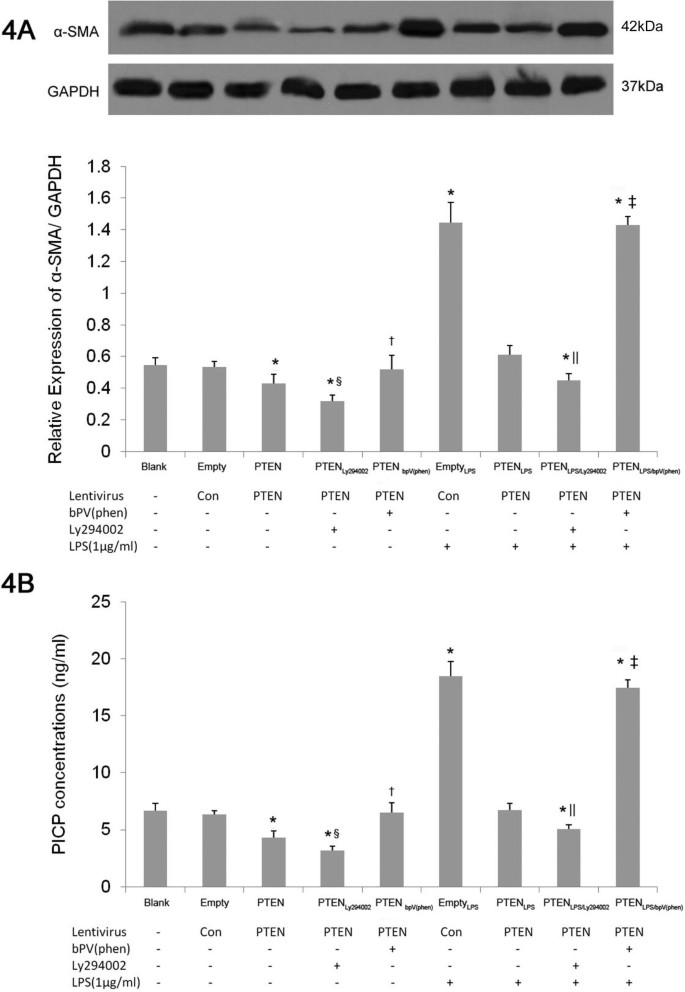
实验目的:评估PTEN过表达对LPS诱导的肺成纤维细胞分化及胶原分泌的影响。方法细节:采用蛋白质免疫印迹检测α-平滑肌肌动蛋白(α-SMA,成纤维细胞向肌成纤维细胞分化的特异性标志物)的蛋白表达;采用ELISA检测细胞培养上清中I型前胶原C端肽(PICP,I型胶原分泌的特异性标志物)的含量。结果解读:与空载体组相比,空载体+LPS组的α-SMA表达及PICP含量显著升高(P<0.05,n=3);与空载体+LPS组相比,PTEN过表达+LPS组的α-SMA表达及PICP含量显著降低(P<0.05,n=3);PTEN抑制剂bpV(phen)可逆转这一抑制作用,PI3-K抑制剂Ly294002可增强抑制效果。实验所用关键产品:α-SMA一抗(Abcam)、PICP ELISA试剂盒(Biorbyt)。
4. Biomarker研究及发现成果解析
本文中涉及的Biomarker为PTEN,作为LPS诱导肺纤维化的潜在治疗靶点Biomarker,其筛选与验证逻辑为“细胞模型验证PTEN表达及活性变化→通路关联分析→功能验证→抑制剂反向验证”的完整链条。PTEN来源于小鼠原代肺成纤维细胞,通过实时荧光定量PCR、蛋白质免疫印迹验证其表达水平,孔雀绿法验证其磷酸酶活性;研究显示,LPS诱导肺成纤维细胞功能异常时,PTEN的表达及磷酸酶活性显著下调,而过表达PTEN可显著逆转LPS诱导的细胞增殖、分化及胶原分泌;未提供PTEN作为Biomarker的特异性与敏感性数据,但通过抑制剂实验证实其磷酸酶活性是调控细胞功能的核心因素。核心成果提炼:PTEN可作为LPS诱导肺纤维化的潜在治疗靶点,其过表达及磷酸酶活性增强可通过抑制PI3-K-Akt-GSK3β通路,显著抑制LPS诱导的肺成纤维细胞增殖(细胞活力降低,S期细胞比例减少,P<0.05,n=3)、分化(α-SMA表达降低,P<0.05,n=3)及胶原分泌(PICP含量降低,P<0.05,n=3);首次证实PTEN的磷酸酶活性比其表达水平在调控肺成纤维细胞功能中更为关键,为肺纤维化的精准靶向治疗提供了新的分子依据,也为后续PTEN相关药物的研发指明了方向。