1. 领域背景与文献引入
文献英文标题:Aldose reductase deficiency inhibits LPS-induced M1 response in macrophages by activating autophagy;发表期刊:Cell & Bioscience;影响因子:未公开;研究领域:巨噬细胞极化与炎症调控。
巨噬细胞是固有免疫系统的核心效应细胞,具有高度可塑性,可根据微环境信号极化为促炎表型(M1型)和抗炎表型(M2型),其中M1极化介导的炎症反应在感染性疾病、自身免疫病、神经退行性疾病等多种病理过程中发挥关键作用。领域共识:脂多糖(LPS)是诱导巨噬细胞M1极化的经典刺激因子,其通过激活Toll样受体4(TLR4)/核因子κB(NF-κB)信号通路,诱导诱导型一氧化氮合酶(iNOS)等促炎介质的表达,进而驱动炎症反应。醛糖还原酶(AR)是多元醇通路的限速酶,近年研究发现其不仅参与糖代谢调控,还在炎症信号转导中发挥重要作用,已有动物模型研究显示AR过表达会加重脓毒症等炎症性疾病的病理损伤,AR抑制剂可缓解过敏性气道炎症、自身免疫性葡萄膜炎等疾病的症状,但AR调控巨噬细胞极化的具体分子机制尚未明确,尤其是AR与自噬通路、NF-κB信号轴的交互作用研究存在空白。本研究正是针对这一核心问题,旨在揭示AR缺失通过自噬调控NF-κB通路进而抑制M1极化的分子机制,为炎症性疾病的治疗提供新的潜在靶点。
2. 文献综述解析
作者从巨噬细胞极化的功能与调控、AR在炎症中的作用、自噬与NF-κB通路的交互作用三个维度对领域内现有研究进行系统性评述,明确了现有研究的进展与未解决的核心问题。
在巨噬细胞极化领域,现有研究已明确M1/M2极化的表型特征与核心调控通路,LPS诱导的TLR4/NF-κB通路是M1极化的核心信号轴,iNOS、CD86是M1极化的关键标志物,但不同病理背景下巨噬细胞极化的精细调控机制仍需深入挖掘,尤其是细胞内代谢酶对极化的调控作用研究较少。在AR与炎症的研究中,已有研究证实AR参与多种炎症模型的调控,其过表达会增强炎症信号传导,抑制AR可缓解炎症反应,但AR调控巨噬细胞极化的直接证据不足,且具体分子机制未阐明。在自噬与NF-κB通路的交互作用领域,现有研究显示自噬可通过降解通路关键组分负调控NF-κB信号,但选择性自噬靶向NF-κB通路核心复合物(如IKK复合物)的研究较少,尤其是AR缺失是否通过自噬调控IKK复合物的机制尚未报道。
本研究的创新价值在于通过对比现有研究的空白,首次揭示AR缺失通过激活选择性自噬降解IKKβ和IKKγ,进而抑制NF-κB通路与M1极化,填补了AR调控巨噬细胞极化机制的空白,为炎症性疾病的治疗提供了新的干预靶点,同时拓展了选择性自噬在巨噬细胞极化精细调控中的作用。
3. 研究思路总结与详细解析
本研究的核心目标是明确AR缺失对LPS诱导巨噬细胞M1极化的调控作用及分子机制,核心科学问题是AR缺失如何通过自噬通路调控NF-κB信号轴,技术路线遵循“表型观察-机制探究-功能验证”的闭环逻辑:首先验证LPS对巨噬细胞AR表达的影响,然后通过AR敲除小鼠模型验证AR缺失对M1极化的抑制作用,接着探究AR缺失对自噬激活的调控,最后明确自噬降解IKK复合物的具体分子机制。
3.1 LPS对巨噬细胞AR表达的调控验证
实验目的:明确LPS刺激是否影响巨噬细胞中AR的表达水平,为后续探究AR对M1极化的调控提供基础。
方法细节:分离野生型(WT)C57BL/6J小鼠的骨髓来源巨噬细胞(BMMs),培养至成熟后用500ng/ml LPS处理24h,采用免疫荧光染色标记AR,通过实时荧光定量PCR(qRT-PCR)检测AR mRNA水平,通过蛋白质免疫印迹(WB)检测AR蛋白水平。
结果解读:免疫荧光结果显示,LPS处理后BMMs中AR阳性灶数量显著增加(n=3,P<0.01);qRT-PCR和WB结果显示,AR的mRNA和蛋白水平均显著上调(n=3,P<0.05),表明LPS可诱导巨噬细胞AR表达升高,提示AR可能参与LPS介导的M1极化过程。
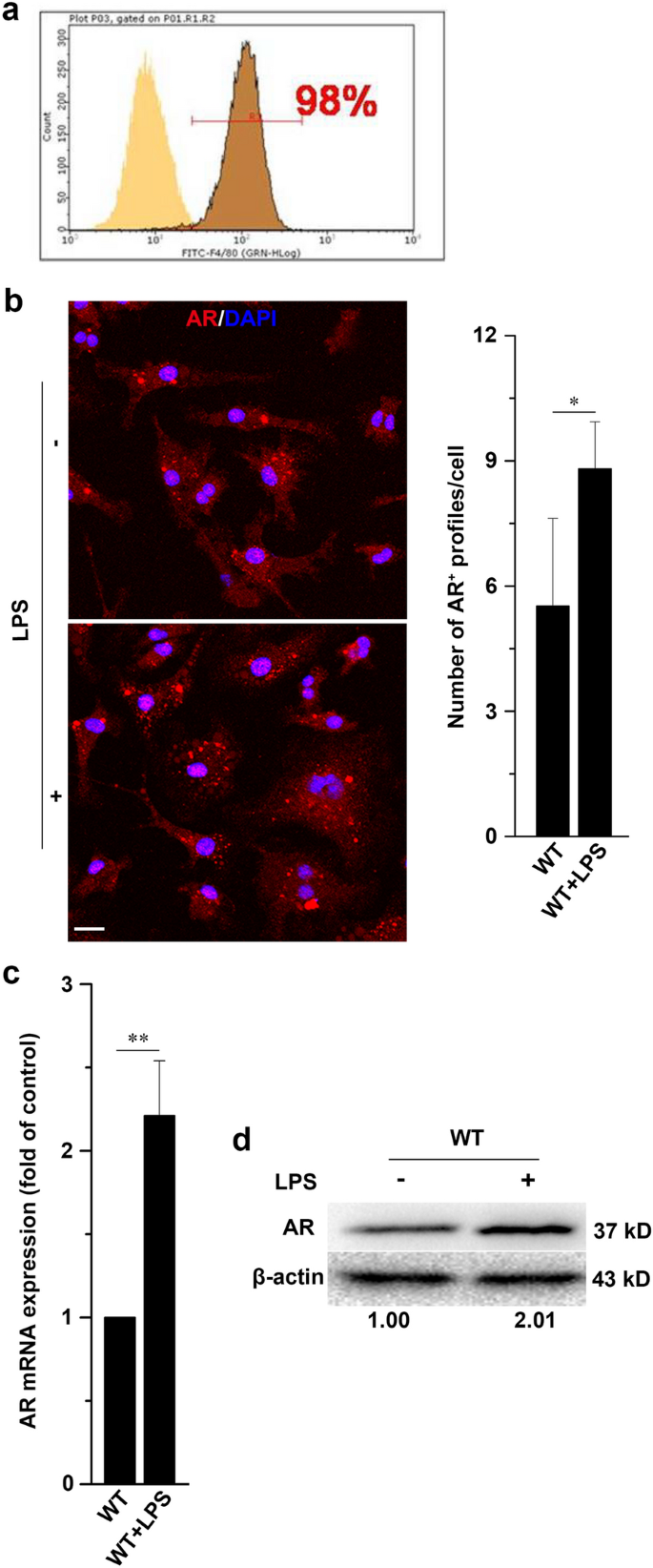
实验所用关键产品:FITC标记的抗F4/80抗体(AbD Serotec)、抗AR抗体(Santa Cruz Biotechnology)、Bio-Rad CFX 96™实时荧光定量PCR系统、Bio-Rad ChemiDoc™ XRS+成像系统等。
3.2 AR缺失对LPS诱导M1极化及NF-κB通路的影响
实验目的:验证AR缺失是否抑制LPS诱导的巨噬细胞M1极化及NF-κB通路激活,明确AR与M1极化的关联。
方法细节:分离WT和AR敲除(KO)小鼠的BMMs,用500ng/ml LPS处理不同时间(0-32h),采用WB检测iNOS、IKKβ、IKKγ、磷酸化IKKα/β(p-IKKα/β)、磷酸化IκBα(p-IκBα)、磷酸化p65(p-p65)等蛋白水平,通过qRT-PCR检测iNOS、CD86等M1标志物及IKKβ、IKKγ的mRNA水平。
结果解读:WB结果显示,LPS处理后WT小鼠BMMs中iNOS蛋白水平随时间升高,16h达到峰值,而AR KO小鼠BMMs中iNOS的诱导显著降低(n=3,P<0.01);同时AR KO小鼠BMMs中p-IKKα/β、IKKβ、IKKγ、p-IκBα、p-p65蛋白水平显著低于WT组(n=3,P<0.05);qRT-PCR结果显示,AR KO小鼠BMMs中IKKβ、IKKγ的mRNA水平与WT组无显著差异,但iNOS、CD86的mRNA水平显著低于WT组(n=3,P<0.01),表明AR缺失通过转录后调控IKKβ和IKKγ蛋白水平,进而抑制NF-κB通路及M1极化。
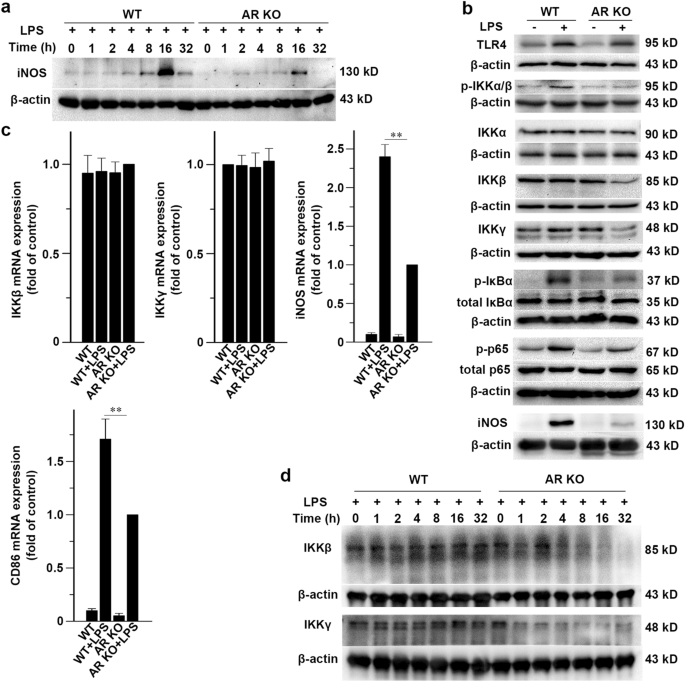
实验所用关键产品:抗iNOS抗体(Abcam)、抗IKKβ抗体(Epitomics)、抗IKKγ抗体(Santa Cruz Biotechnology)等。
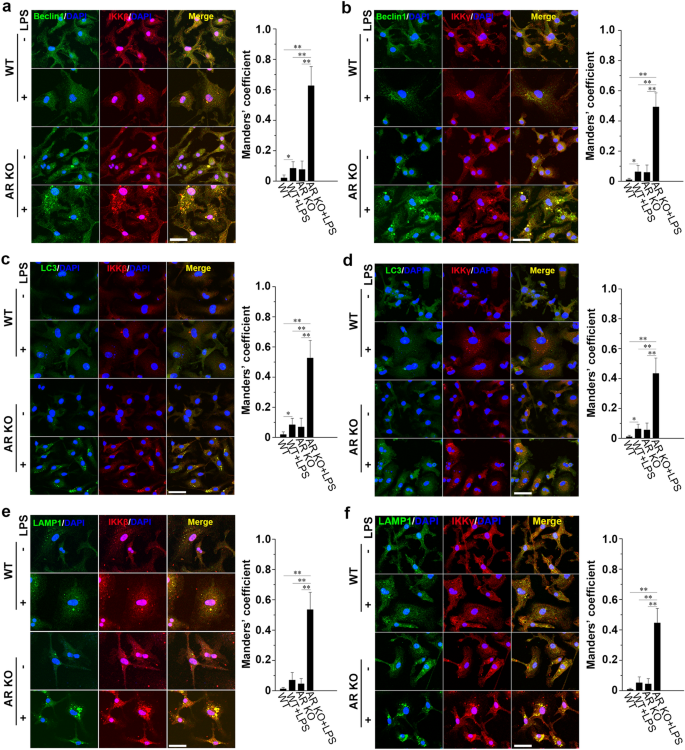
3.3 AR缺失对巨噬细胞自噬的调控作用
实验目的:探究AR缺失是否影响LPS诱导的巨噬细胞自噬激活及自噬体成熟,明确AR与自噬的关联。
方法细节:分离WT和AR KO小鼠的BMMs,用500ng/ml LPS处理16h,采用免疫荧光染色标记beclin1、微管相关蛋白1轻链3β(LC3β)、溶酶体相关膜蛋白1(LAMP1),检测其表达及共定位情况;采用WB检测LC3II/LC3I比值、LAMP1蛋白水平;通过透射电子显微镜观察自噬体和自溶酶体的数量。
结果解读:免疫荧光结果显示,AR KO小鼠BMMs中beclin1阳性灶数量显著多于WT组(n=3,P<0.01),LPS处理后进一步增加;LC3阳性空泡数量在AR KO组显著升高,LC3II/LC3I比值也显著高于WT组(n=3,P<0.05);LAMP1与LC3的共定位率在AR KO组显著升高(n=3,P<0.01);透射电子显微镜结果显示,AR KO组自噬体和自溶酶体数量显著多于WT组,LPS处理后进一步增加,表明AR缺失可促进巨噬细胞自噬体形成及成熟。
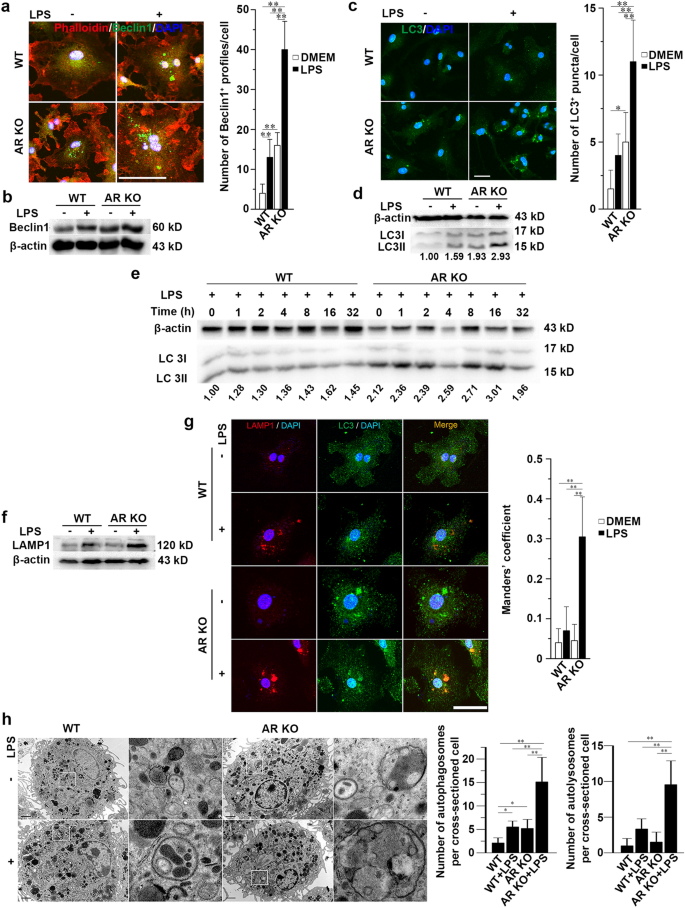
实验所用关键产品:抗beclin1抗体(Santa Cruz Biotechnology)、抗LC3β抗体(Santa Cruz Biotechnology)、抗LAMP1抗体(Santa Cruz Biotechnology)、JEOL JEM-1230透射电子显微镜等。
3.4 自噬在AR缺失调控IKK复合物及M1极化中的功能验证
实验目的:验证自噬是否介导AR缺失对IKK复合物的降解及M1极化的抑制,明确自噬的功能作用。
方法细节:用自噬抑制剂3-甲基腺嘌呤(3-MA,3mM)或溶酶体抑制剂氯化铵(NH4Cl,10mM)处理AR KO小鼠BMMs,预处理后用500ng/ml LPS刺激16h,采用WB检测IKKβ、IKKγ、iNOS蛋白水平,采用免疫荧光检测LC3阳性空泡数量。
结果解读:免疫荧光结果显示,3-MA或NH4Cl处理可显著减少AR KO小鼠BMMs中LC3阳性空泡数量(n=3,P<0.01);WB结果显示,3-MA或NH4Cl处理后,AR KO小鼠BMMs中IKKβ、IKKγ蛋白水平显著恢复(n=3,P<0.05),iNOS的诱导也显著增强(n=3,P<0.01),表明自噬介导了AR缺失对IKK复合物的降解及M1极化的抑制。
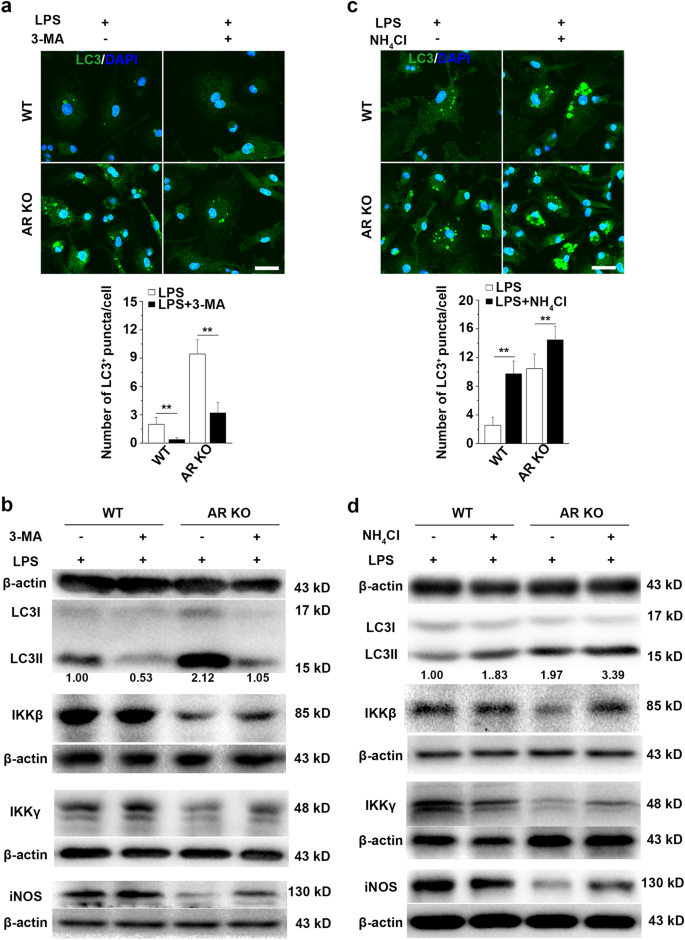
实验所用关键产品:3-MA、NH4Cl(Sigma-Aldrich)等。
3.5 AR缺失介导IKK复合物自噬降解的分子机制
实验目的:明确AR缺失如何通过泛素化和自噬衔接蛋白介导IKK复合物的降解,解析具体分子机制。
方法细节:分离WT和AR KO小鼠的BMMs,用500ng/ml LPS处理1h,采用免疫荧光染色标记IKKβ、IKKγ与beclin1、LC3、LAMP1、K63位多聚泛素链(PolyUb)、p62,检测其共定位情况。
结果解读:免疫荧光结果显示,AR KO组中IKKβ、IKKγ与beclin1、LC3、LAMP1的共定位率显著高于WT组(n=3,P<0.01);同时IKKβ、IKKγ与K63位多聚泛素链、p62的共定位率也显著升高(n=3,P<0.05),表明AR缺失诱导IKKβ、IKKγ发生K63位泛素化,进而被自噬衔接蛋白p62识别并招募至自噬体,最终在溶酶体中降解。
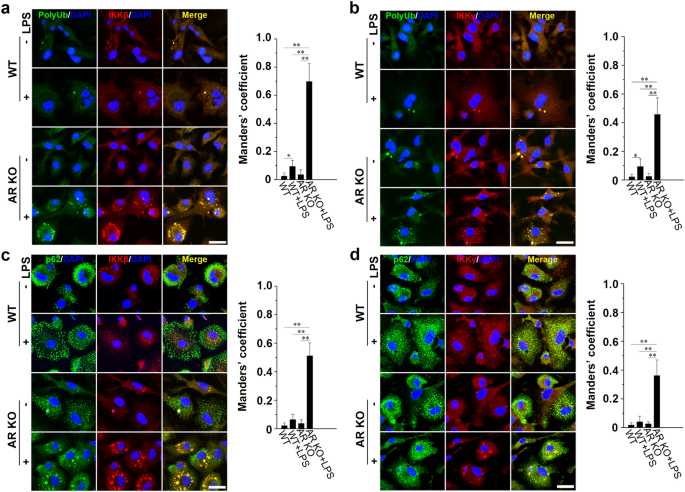
实验所用关键产品:抗K63泛素链抗体(Abcam)、抗p62抗体(Santa Cruz Biotechnology)等。
4. Biomarker研究及发现成果
本研究未聚焦传统疾病诊断或预后Biomarker,而是揭示了醛糖还原酶(AR)作为巨噬细胞M1极化调控的功能性Biomarker,其表达水平与巨噬细胞M1极化程度正相关,为炎症性疾病的治疗提供潜在靶点。
AR作为功能性Biomarker,其表达水平直接影响巨噬细胞M1极化的程度,筛选与验证逻辑为:首先通过LPS刺激验证AR表达上调与M1极化的关联,然后通过AR敲除小鼠模型验证其缺失对M1极化的抑制作用,最后明确其调控机制。AR的来源为巨噬细胞内源性表达,验证方法包括免疫荧光、qRT-PCR、WB检测AR表达水平,通过AR敲除小鼠模型验证其功能,特异性数据显示LPS刺激后WT小鼠BMMs中AR表达显著上调(n=3,P<0.05),AR缺失后M1极化标志物iNOS、CD86的表达显著降低(n=3,P<0.01)。
本研究首次揭示AR缺失通过激活选择性自噬降解IKKβ和IKKγ,进而抑制NF-κB通路及M1极化,明确了AR-自噬-IKK-NF-κB轴在巨噬细胞极化中的调控作用,创新性在于阐明了AR作为炎症调控靶点的分子机制,为炎症性疾病的治疗提供了新的干预策略。本研究未涉及疾病诊断或预后相关的Biomarker研究,因此无相关统计学数据(如风险比HR、ROC曲线AUC等)。