1. 领域背景与文献引入
文献英文标题:Presenilin-1 F105C mutation leads to tau accumulation in human neurons via the Akt/mTORC1 signaling pathway;发表期刊:Cell & Bioscience;影响因子:未公开;研究领域:神经科学-家族性阿尔茨海默病机制研究
阿尔茨海默病(AD)是全球最常见的神经退行性疾病,以进行性记忆丧失和认知障碍为核心特征,其中家族性阿尔茨海默病(FAD)约占AD病例的1%,而早老素-1(PS1)突变是FAD最主要的致病原因,目前已报道超过300种PS1突变,可引发Aβ斑块沉积、tau神经原纤维缠结、突触异常及神经元丢失等典型AD病理特征。哺乳动物雷帕霉素靶蛋白(mTOR)在细胞稳态调控中发挥核心作用,其功能失调已被证实与AD发病密切相关,但PS1突变是否会在人神经元中导致mTOR通路失调,仍是领域内未解决的关键科学问题。
现有研究多依赖PS1突变过表达模型或患者成纤维细胞,难以精准模拟人神经元的病理变化,而诱导多能干细胞(iPSC)技术为AD机制研究提供了更贴近生理的模型,但个体基因组差异限制了其应用。针对中国人群特有的早发FAD致病突变PS1 F105C,目前尚未有其在人神经元中致病机制的报道,因此本研究通过CRISPR/Cas9结合piggyBac转座子系统构建PS1 F105C敲入的等基因iPSC,并分化为神经元,系统解析该突变对mTORC1通路及tau积累的调控作用,填补了PS1突变在人神经元中mTOR调控机制的研究空白,为AD治疗提供了新的潜在靶点。
2. 文献综述解析
作者从PS1突变与FAD的关联、mTOR通路与AD病理的关系、iPSC模型在AD研究中的应用三个维度对领域研究进行了系统梳理。
现有研究表明,PS1突变可通过γ-分泌酶依赖或非依赖途径参与AD病理过程:一方面,PS1作为γ-分泌酶的核心亚基,其突变可改变淀粉样前体蛋白(APP)的加工过程,导致Aβ42/Aβ40比值升高;另一方面,PS1突变还可引发内溶酶体系统和自噬系统功能障碍,影响细胞内蛋白稳态。mTOR通路失调在AD动物模型和患者脑组织中均有观察到,其下游靶点参与tau蛋白磷酸化和自噬调控,而自噬功能异常是tau蛋白清除障碍的重要原因。iPSC模型能够模拟AD的病理特征,但个体基因组背景差异会干扰实验结果,通过基因编辑构建的等基因iPSC模型可有效排除该干扰,但传统基因编辑方法构建纯合突变iPSC的效率极低,限制了其广泛应用。
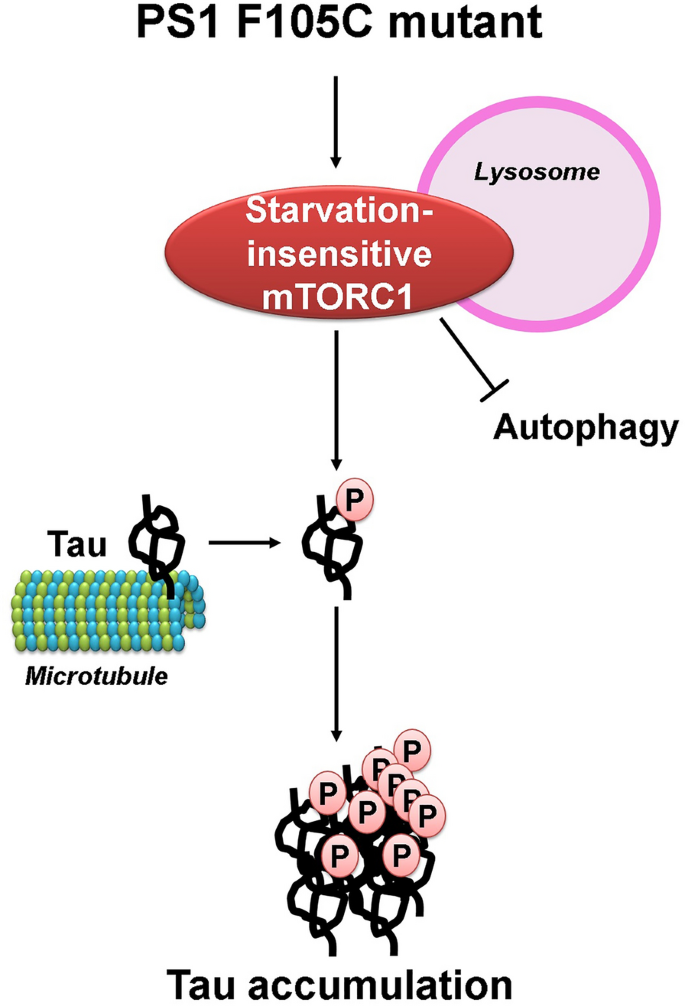
现有研究的局限性在于,针对中国人群特有的PS1 F105C突变的致病机制研究仍为空白,且尚未明确PS1突变是否会在人神经元中直接导致mTORC1通路失调。本研究的创新点在于,首次结合CRISPR/Cas9与piggyBac转座子系统,高效构建了无外源序列残留的PS1 F105C杂合和纯合敲入等基因iPSC神经元模型,在人神经元水平明确了PS1 F105C突变通过Akt/mTORC1通路失调导致tau蛋白积累的分子机制,为AD的mTOR靶向治疗提供了直接的实验证据。
3. 研究思路总结与详细解析
本研究的核心目标是明确PS1 F105C突变对人神经元中mTORC1通路活性及tau蛋白积累的影响,核心科学问题是PS1 F105C突变是否通过mTORC1通路失调介导tau蛋白病理,技术路线遵循“基因编辑构建模型→细胞分化与表型检测→通路机制解析→治疗靶点验证”的闭环逻辑。
3.1 等基因PS1 F105C敲入iPSC构建与验证
实验目的是构建无外源序列残留的PS1 F105C杂合和纯合敲入iPSC,排除个体基因组差异对实验结果的干扰。方法细节:从28岁健康中国女性尿液细胞重编程得到UC-H2-iPSCs,采用CRISPR/Cas9结合piggyBac转座子系统进行两步基因编辑:第一步在PSEN1等位基因1插入携带puro-TK筛选标记的piggyBac载体,通过嘌呤霉素筛选获得杂合突变克隆,再转染转座酶并经FIAU处理去除载体序列;第二步在杂合突变细胞的等位基因2插入携带HygR-TK筛选标记的piggyBac载体,经潮霉素筛选获得纯合突变克隆,同样去除载体序列;通过Sanger测序、核型分析、多能性标记免疫荧光染色(Oct4)及畸胎瘤形成实验验证iPSC的基因型和多能性。结果解读:成功构建了PS1 F105C杂合(18.9%克隆效率)和纯合(13.7%克隆效率)敲入iPSC,所有细胞系核型正常,多能性标记表达正常,且未检测到脱靶效应。
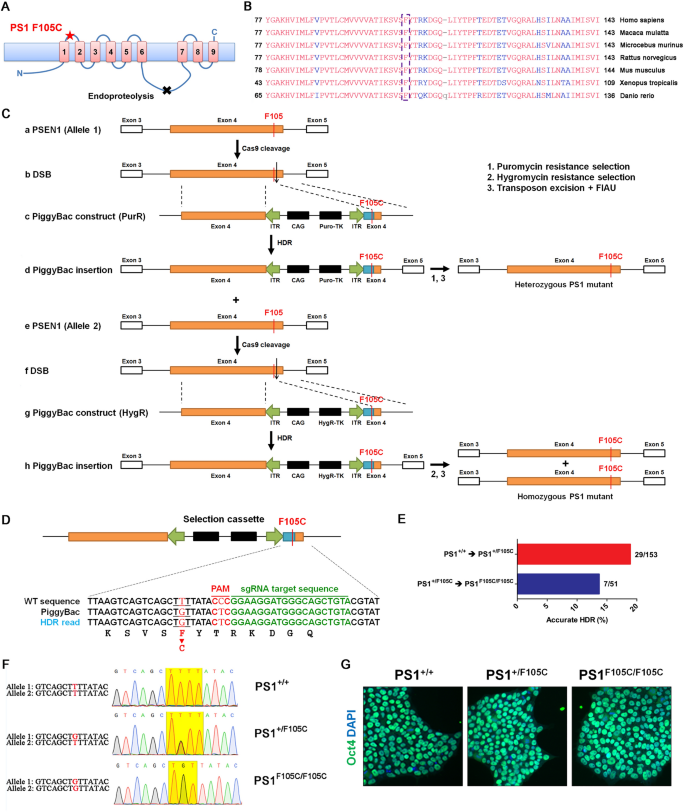
产品关联:实验所用关键产品:CRISPR/Cas9系统、piggyBac转座子载体(Dr. Yuet Wai Kan馈赠)、Matrigel(BD Biosciences)、mTeSR1培养基(STEMCELL Technologies)、嘌呤霉素、潮霉素、FIAU等,其余未明确品牌的试剂为领域常规使用的iPSC重编程、基因编辑及细胞培养试剂。
3.2 iPSC分化为神经元及Aβ水平检测
实验目的是验证PS1 F105C突变对神经元分化能力及Aβ产生的影响。方法细节:将iPSC分化为神经干细胞(NSCs),再进一步分化为成熟神经元(培养20天);采用免疫荧光染色检测NSCs标记(SOX2、Nestin)和神经元标记(TUJ1)的表达,Western blotting检测PS1剪切片段(PS1-NTF、PS1-CTF)和全长APP的水平,ELISA检测神经元培养上清中Aβ40、Aβ42的含量。结果解读:所有iPSC系分化为NSCs和成熟神经元的能力一致,PS1 F105C突变不影响PS1的内蛋白水解剪切过程,但全长APP的水平随突变拷贝数增加而显著升高;突变神经元培养上清中Aβ40、Aβ42的水平及Aβ42/Aβ40比值均显著高于对照神经元(n=4,P<0.01或P<0.005),与AD动物模型和患者的病理特征一致。
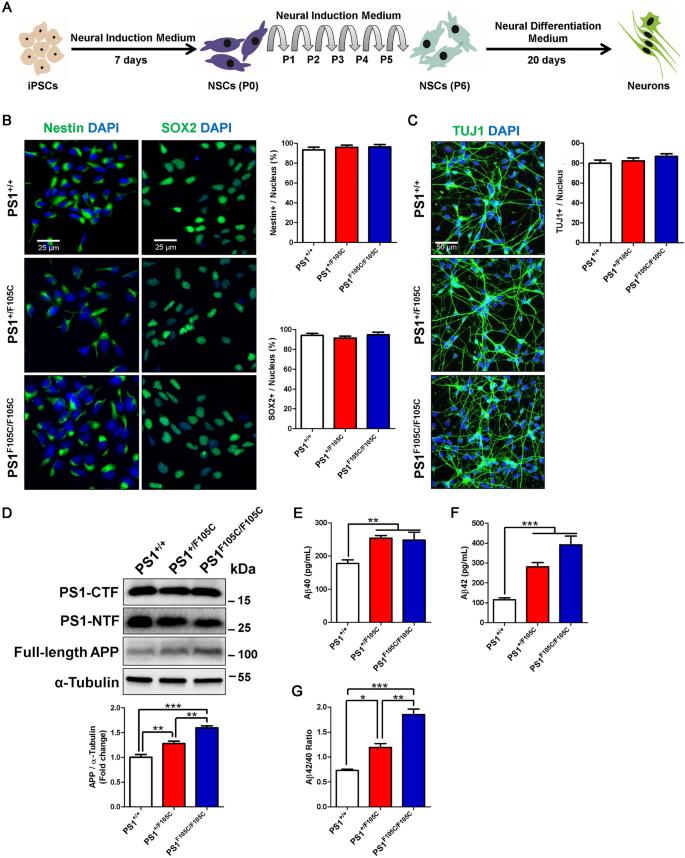
产品关联:实验所用关键产品:神经诱导培养基(Life Technologies)、N2B27培养基、GDNF、BDNF、NT3(PeproTech)、Aβ40/Aβ42 ELISA试剂盒(Wako)、免疫荧光抗体(SOX2、Nestin、TUJ1)、Western blotting抗体(PS1-NTF、PS1-CTF、APP)等。
3.3 PS1 F105C神经元中mTORC1通路活性分析
实验目的是明确PS1 F105C突变对mTORC1通路饥饿响应能力的影响。方法细节:将神经元用Earle"s平衡盐溶液(EBSS)饥饿处理4小时,通过Western blotting检测Akt、mTOR、p70S6K的磷酸化水平,免疫荧光染色检测mTOR与溶酶体标记LAMP1的共定位情况。结果解读:在正常喂养条件下,突变与对照神经元的mTOR通路磷酸化蛋白水平无显著差异;但在饥饿条件下,对照神经元中mTOR、p70S6K、Akt的磷酸化水平显著降低,而PS1 F105C突变神经元中这些蛋白的磷酸化水平仍维持在较高水平(n=3,P<0.005);免疫荧光结果显示,对照神经元在饥饿时mTOR从溶酶体膜扩散至细胞质,而突变神经元中大部分mTOR仍与LAMP1共定位(n≥30,P<0.005),表明PS1 F105C突变导致mTORC1通路对饥饿的响应性降低。
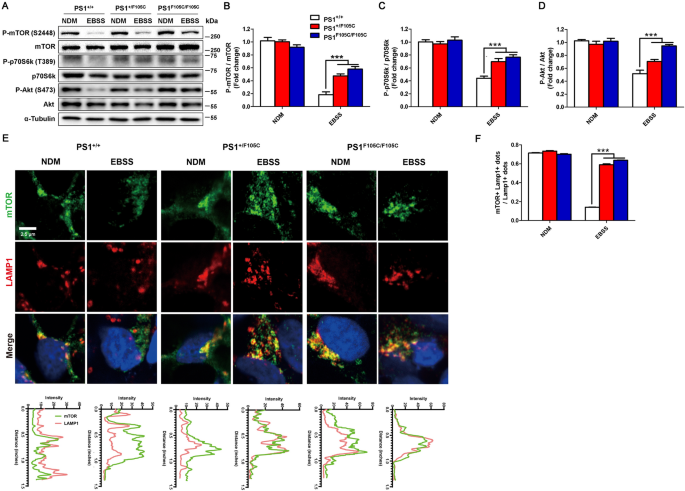
产品关联:实验所用关键产品:EBSS、Western blotting抗体(p-mTOR、T-mTOR、p-p70S6K、T-p70S6K、p-Akt、T-Akt)、免疫荧光抗体(mTOR、LAMP1)等。
3.4 PS1 F105C神经元自噬功能评估
实验目的是解析PS1 F105C突变对自噬不同阶段功能的影响。方法细节:通过Western blotting检测自噬体标记LC3-II的水平,结合氯喹(CQ)处理评估自噬流;免疫荧光染色检测LC3与LAMP1的共定位情况;采用LysoTracker Red染色检测溶酶体pH,DQ-Red BSA染色检测溶酶体降解功能;Western blotting检测自噬底物p62的残留水平。结果解读:饥饿可诱导所有神经元的LC3-II水平升高,但突变神经元的LC3-II水平显著高于对照;CQ处理后,突变神经元的基础自噬流和饥饿诱导的自噬流均显著低于对照神经元(n=3,P<0.05或P<0.005);免疫荧光结果显示,对照神经元在饥饿时LC3与LAMP1大量共定位,而突变神经元中两者共定位极少;LysoTracker Red和DQ-Red BSA染色结果显示,突变神经元的溶酶体pH和降解功能均正常,但饥饿时突变神经元的p62残留水平显著高于对照(n=3,P<0.005),表明PS1 F105C突变导致自噬体-溶酶体融合障碍,进而引发自噬功能异常。
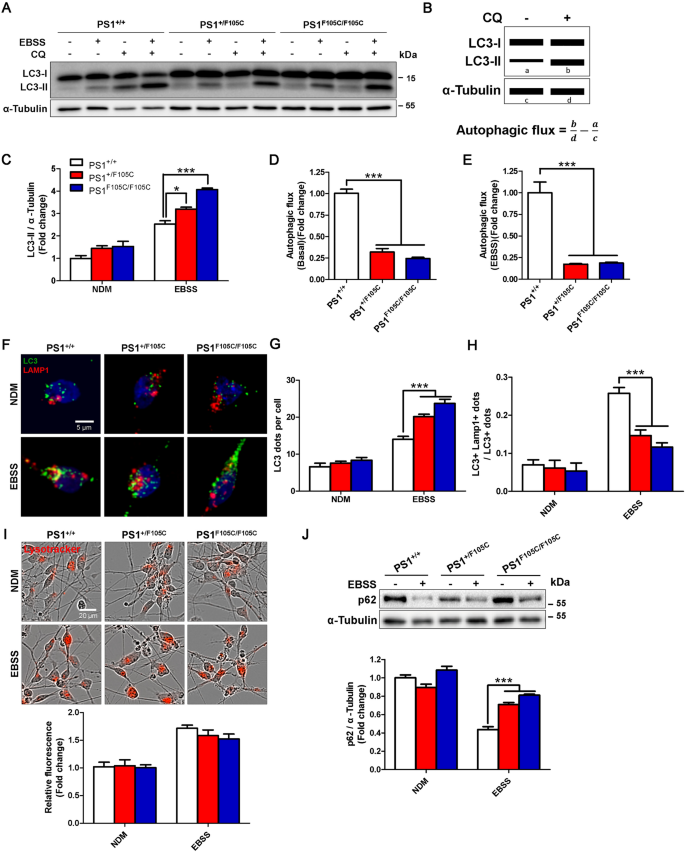
产品关联:实验所用关键产品:氯喹、LysoTracker Red DND-99(Invitrogen)、DQ-Red BSA(Molecular Probes/Invitrogen)、Western blotting抗体(LC3、p62)等。
3.5 mTOR抑制剂对tau积累的改善作用验证
实验目的是验证mTOR通路抑制是否能清除PS1 F105C突变神经元中的tau蛋白积累。方法细节:将PS1 F105C纯合突变神经元用1μM Torin1处理2天,通过Western blotting检测总tau和磷酸化tau(At8)的水平,免疫荧光染色检测tau蛋白在神经元胞体的积累情况。结果解读:PS1 F105C突变神经元的总tau水平、磷酸化tau/tau比值显著高于对照神经元(n=3,P<0.05或P<0.005),且tau蛋白在胞体的积累量随突变拷贝数增加而升高(n≥50,P<0.005);Torin1处理后,突变神经元中Akt、p70S6K的磷酸化水平显著降低,总tau水平、磷酸化tau/tau比值显著下降(n=3,P<0.005),胞体tau蛋白积累量也显著减少(n≥50,P<0.005),表明mTOR抑制剂可有效改善PS1 F105C突变神经元的tau病理。
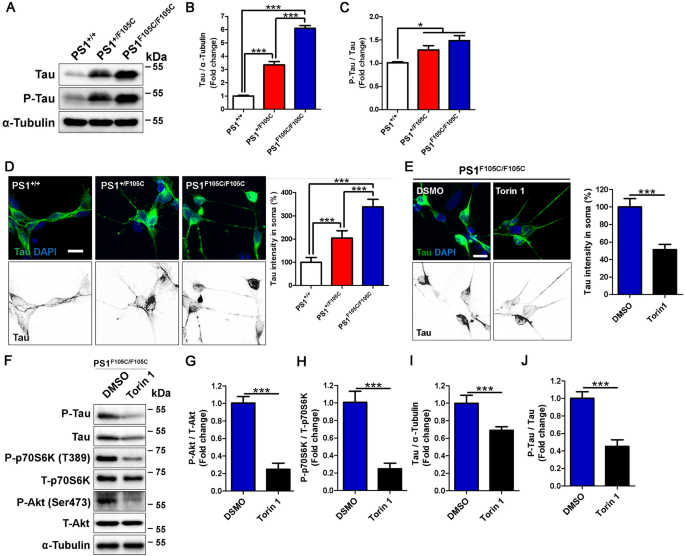
产品关联:实验所用关键产品:Torin1、Western blotting抗体(tau、p-tau(At8))、免疫荧光抗体(tau)等。
4. Biomarker研究及发现成果解析
Biomarker定位
本研究涉及的Biomarker包括病理Biomarker(Aβ40、Aβ42、磷酸化tau(At8))和通路Biomarker(p-mTOR、p-p70S6K、p-Akt),筛选与验证逻辑为:基于PS1 F105C敲入的等基因神经元模型,通过细胞水平实验验证这些Biomarker与突变的关联及通路调控关系。
研究过程详述
Aβ40、Aβ42来源于神经元培养上清,采用ELISA定量检测,结果显示PS1 F105C突变神经元中Aβ40、Aβ42水平及Aβ42/Aβ40比值显著升高(n=4,P<0.01或P<0.005);磷酸化tau(At8)和总tau通过Western blotting和免疫荧光检测,突变神经元中总tau水平升高,磷酸化tau/tau比值显著升高(n=3,P<0.05或P<0.005),tau蛋白在胞体的积累量显著增加(n≥50,P<0.005);mTOR通路磷酸化蛋白通过Western blotting检测,饥饿条件下突变神经元中p-mTOR、p-p70S6K、p-Akt水平显著高于对照神经元(n=3,P<0.005)。本研究未进行临床样本验证,细胞水平实验显示这些Biomarker能够有效区分突变与对照神经元,具有良好的特异性。
核心成果提炼
首次在人神经元中明确了PS1 F105C突变通过Akt/mTORC1通路失调导致tau蛋白积累的分子机制,其中磷酸化tau(At8)可作为PS1 F105C突变神经元的病理Biomarker,mTOR通路磷酸化蛋白可作为通路失调的Biomarker。研究还发现mTOR抑制剂Torin1可有效降低tau蛋白积累,提示mTOR通路是AD治疗的潜在靶点。本研究的创新性在于,针对中国人群特有的早发FAD致病突变PS1 F105C,在人神经元水平揭示了其致病机制,为该类FAD的诊断和治疗提供了新的科学依据。