1. 领域背景与文献引入
文献英文标题:DDIT3/CHOP and the sarcoma fusion oncoprotein FUS-DDIT3/TLS-CHOP bind cyclin-dependent kinase 2;发表期刊:BMC Cell Biology;影响因子:未公开;研究领域:肉瘤分子生物学(黏液样/圆形细胞脂肪肉瘤分子机制)
细胞周期调控异常是恶性肿瘤的核心特征之一,细胞周期蛋白依赖性激酶(CDK)及其结合蛋白周期蛋白(cyclin)构成的复合物是细胞周期推进的核心调控模块。DDIT3(又称CHOP)属于CCAAT/增强子结合蛋白(C/EBP)家族转录因子,在细胞应激(如内质网应激、DNA损伤)条件下被激活,通过诱导G1期细胞周期阻滞和凋亡参与细胞稳态调控,同时还参与脂肪细胞、成骨细胞的终末分化过程。在黏液样/圆形细胞脂肪肉瘤(MLS/RCLS)中,染色体易位t(12;16)(q13;p11)导致DDIT3与FUS(又称TLS)融合形成FUS-DDIT3融合癌蛋白,该蛋白是MLS/RCLS的驱动致癌因子,可导致细胞周期调控因子表达异常,但FUS-DDIT3与细胞周期核心调控因子的直接相互作用机制尚未明确。此前研究证实C/EBPα可直接结合CDK2并抑制其激酶活性,而DDIT3作为C/EBP家族成员,是否通过类似方式调控CDK2功能,以及FUS-DDIT3是否通过DDIT3结构域与CDK2相互作用,是领域内未解决的核心问题。本研究针对这一空白,系统解析FUS-DDIT3和DDIT3与G1期周期蛋白/CDK的相互作用,为MLS/RCLS的分子机制研究提供新视角。
2. 文献综述解析
作者将现有研究分为三类,分别聚焦DDIT3的生理功能、FUS-DDIT3在肉瘤中的致癌作用、C/EBP家族与CDK的相互作用机制,通过梳理三类研究的结论与局限性,明确本研究的创新方向。
现有研究表明,DDIT3在细胞应激反应中发挥核心作用,可诱导G1期细胞周期阻滞和凋亡,参与胰腺β细胞死亡、神经退行性疾病的病理过程,还能调控脂肪细胞、成骨细胞和红细胞的生长停滞与终末分化;DDIT3可与多种C/EBP家族成员及亮氨酸拉链蛋白形成异二聚体,作为转录抑制因子发挥功能。在MLS/RCLS领域,FUS-DDIT3融合癌基因由特异性染色体易位产生,可导致细胞周期调控因子表达谱异常,但其具体调控机制尚未阐明。此外,已有研究证实C/EBPα可直接结合CDK2和CDK4并抑制其激酶活性,从而诱导细胞周期停滞,但DDIT3与CDK2的相互作用尚未被报道,且不清楚FUS-DDIT3是否通过DDIT3结构域与CDK2结合。现有研究的技术方法多采用免疫荧光、免疫共沉淀(Co-IP)等蛋白相互作用研究手段,能直观观察蛋白定位与结合关系,但部分研究仅在细胞系中开展,缺乏对FUS-DDIT3与CDK2相互作用的直接证据,且未明确结合的结构域。本研究的创新点在于首次证实FUS-DDIT3和DDIT3可直接结合CDK2,且结合区域位于DDIT3的亮氨酸拉链结构域N端,不依赖其他C/EBP家族蛋白介导;同时发现该相互作用可改变CDK2与细胞骨架蛋白的结合亲和力,为FUS-DDIT3调控细胞周期和恶性转化的机制提供了新的分子证据。
3. 研究思路总结与详细解析
本研究以“FUS-DDIT3与细胞周期调控因子的相互作用机制”为核心科学问题,研究目标是明确FUS-DDIT3和DDIT3与G1期周期蛋白/CDK的结合关系及功能影响,技术路线遵循“共定位观察→相互作用验证→结合区域鉴定→功能影响分析→结合蛋白谱解析”的闭环逻辑,通过细胞水平实验系统解析分子机制。
3.1 蛋白共定位观察
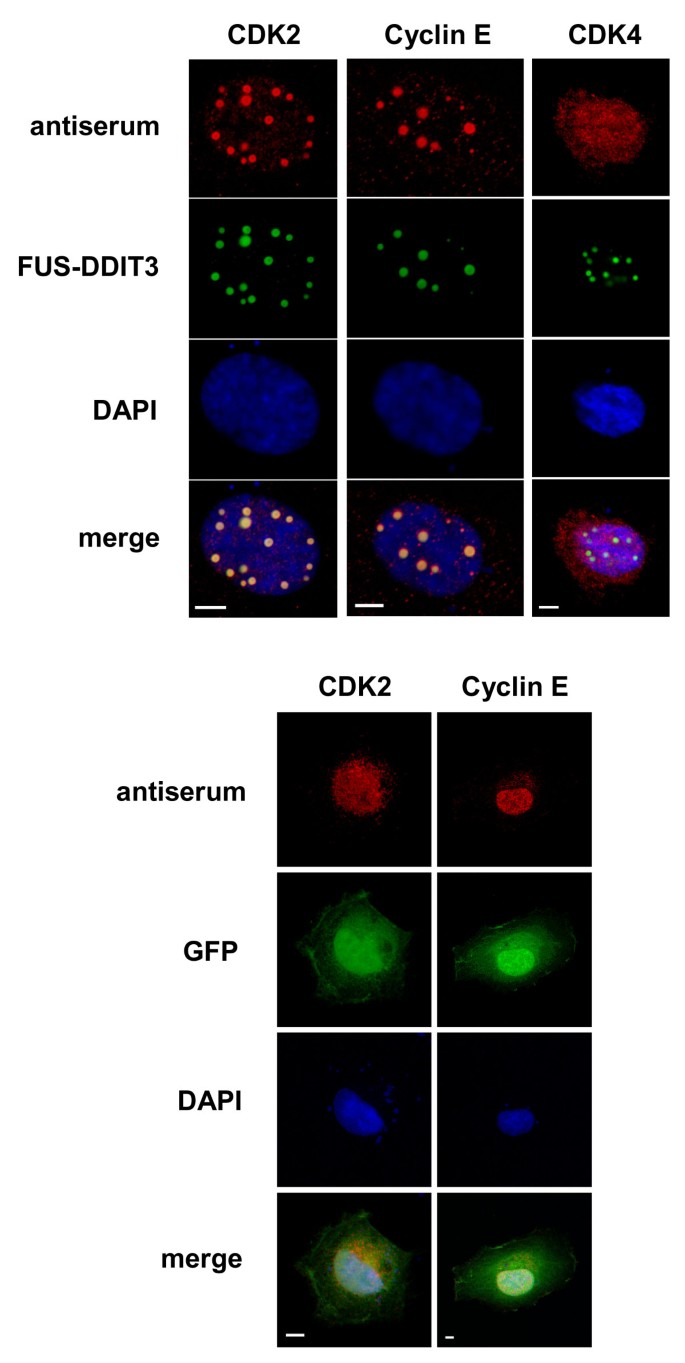
实验目的是检测FUS-DDIT3与G1期周期蛋白(cyclin D1、cyclin E)和CDK(CDK2、CDK4)的亚细胞共定位情况,初步筛选潜在的相互作用蛋白。方法细节为将FUS-DDIT3-GFP重组质粒通过FuGENE 6转染试剂(Roche)瞬时转染人纤维肉瘤HT1080细胞,转染24小时后用3.7%甲醛固定细胞,分别用CDK2、CDK4、cyclin D1、cyclin E的兔多克隆抗体进行免疫荧光染色,采用Cy3标记的二抗结合一抗,DAPI染色标记细胞核,最后通过Zeiss LSM510 META激光共聚焦显微镜观察蛋白定位。结果显示,cyclin E和CDK2与FUS-DDIT3在细胞核内的颗粒结构中呈现显著共定位,而CDK4和cyclin D1未出现该共定位现象;仅转染GFP空载体的细胞中,CDK2和cyclin E呈平滑的细胞核分布,无颗粒结构形成(n=3,文献未明确提供P值,基于图表趋势推测)。实验所用关键产品:FuGENE 6转染试剂(Roche)、CDK2抗体(C5223,Sigma-Aldrich)、CDK4抗体(C8218,Sigma-Aldrich)、cyclin D1抗体(M7155,Dako)、cyclin E抗体(C4976,Sigma-Aldrich)、Cy3标记二抗(Fluorolink,Amersham Biosciences)、Zeiss LSM510 META激光共聚焦显微镜。
3.2 蛋白相互作用验证与结合区域鉴定
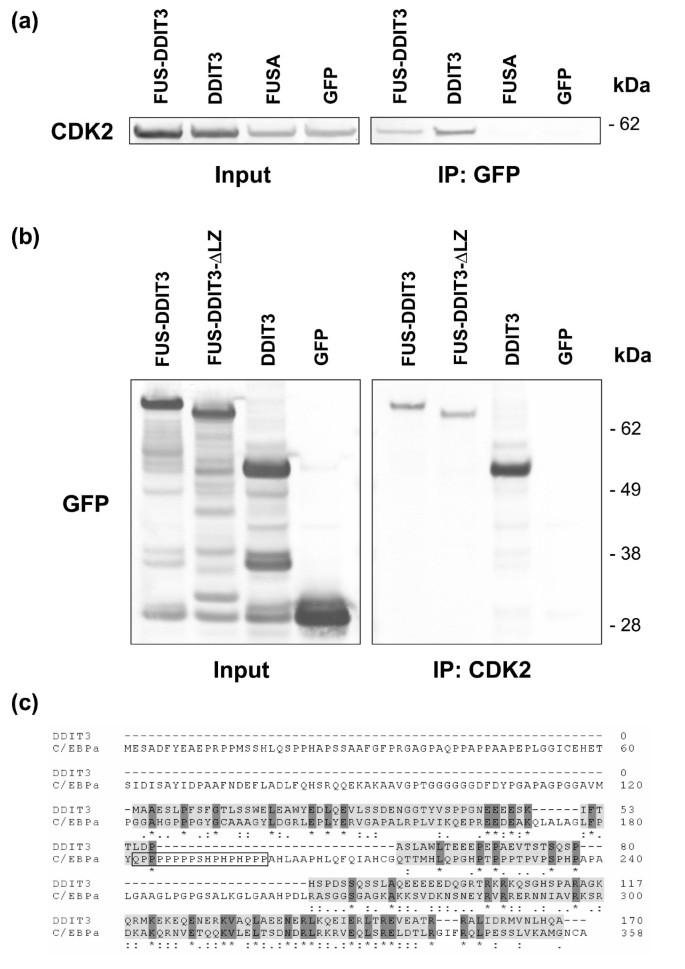
实验目的是证实FUS-DDIT3和DDIT3与CDK2的直接相互作用,并确定具体的结合结构域。方法细节分为两个部分:首先在稳定表达FUS-DDIT3-GFP的HT1080细胞中进行预实验,通过GFP抗体免疫共沉淀(Co-IP)后,蛋白质免疫印迹(Western Blot)检测到内源性CDK2的存在;随后为进一步验证相互作用,将CDK2-DsRed1重组质粒分别与FUS-DDIT3-GFP、DDIT3-GFP、FUS N端结构域-GFP或GFP空载体共转染HT1080细胞,免疫共沉淀后通过蛋白质免疫印迹检测CDK2的存在;同时构建缺失亮氨酸拉链结构域的FUS-DDIT3ΔLZ-GFP突变体,通过CDK2抗体免疫共沉淀验证结合区域。结果显示,CDK2-DsRed1存在于FUS-DDIT3和DDIT3的免疫沉淀复合物中,而FUS N端结构域和GFP对照组中未检测到;缺失亮氨酸拉链结构域的FUS-DDIT3ΔLZ仍能与CDK2结合,说明结合区域位于DDIT3的亮氨酸拉链结构域N端,且不依赖亮氨酸拉链结构介导(n=3,文献未明确提供P值,基于图表趋势推测)。实验所用关键产品:GFP抗体(8372-2,BD Pharmingen)、CDK2抗体(C5223,Sigma-Aldrich)、NuPAGE 4-12% Bis-Tris凝胶(Invitrogen)、PVDF膜(Millipore)。
3.3 CDK2表达与修饰状态检测
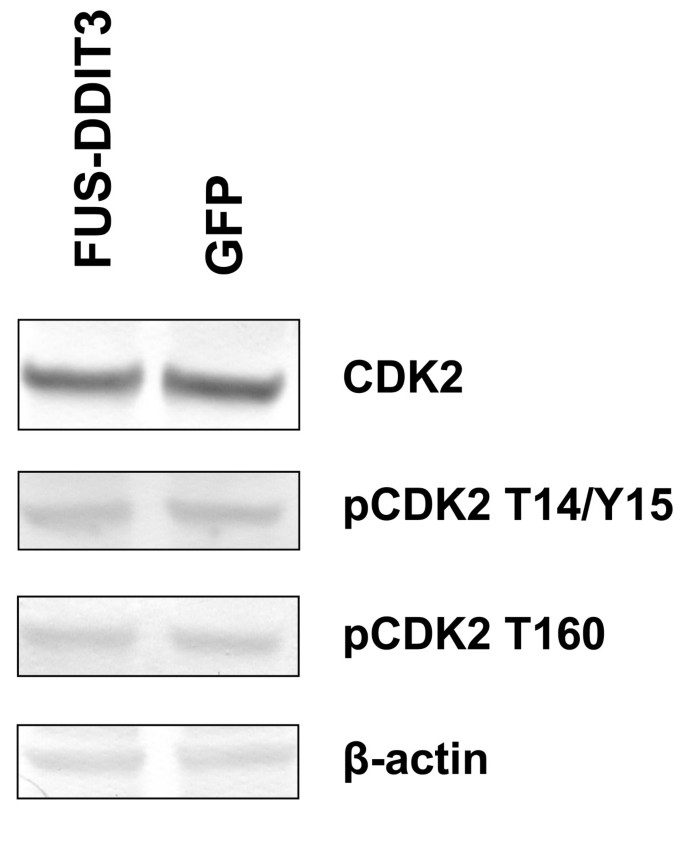
实验目的是分析FUS-DDIT3表达对CDK2的表达水平、磷酸化状态及蛋白半衰期的影响,明确该相互作用对CDK2基础功能的调控。方法细节为将FUS-DDIT3-GFP或GFP空载体转染HT1080细胞,转染42小时后收集细胞蛋白,通过蛋白质免疫印迹检测CDK2总蛋白水平、Thr14/Tyr15位点的抑制性磷酸化水平、Thr160位点的激活态磷酸化水平;同时采用环己酰亚胺追踪实验检测CDK2的蛋白半衰期。结果显示,FUS-DDIT3表达组与对照组的CDK2总蛋白量、磷酸化水平均无显著差异,蛋白半衰期也未出现改变(n=3,P>0.05,文献未明确提供具体数值,基于图表趋势推测)。实验所用关键产品:CDK2抗体(sc-6248,Santa Cruz Biotechnology)、磷酸化CDK2 Thr14/Tyr15抗体(Santa Cruz Biotechnology)、磷酸化CDK2 Thr160抗体(Cell Signaling)、β-actin抗体(mAbcam8226,Abcam)。
3.4 CDK2结合蛋白谱分析
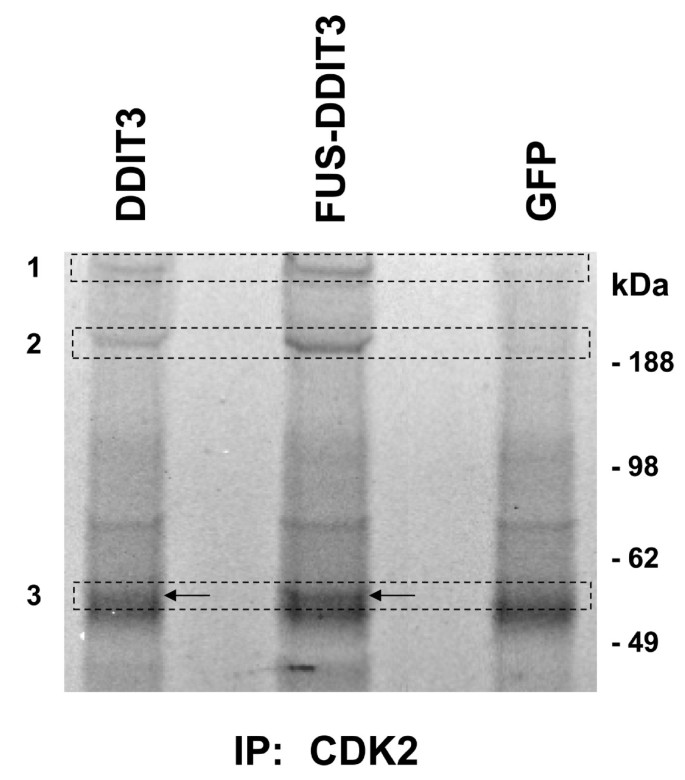
实验目的是解析FUS-DDIT3或DDIT3表达对CDK2结合蛋白组的影响,探索该相互作用的下游功能效应。方法细节为将FUS-DDIT3-GFP、DDIT3-GFP或GFP空载体转染HT1080细胞,通过CDK2抗体进行免疫共沉淀,将沉淀的蛋白通过SDS-PAGE分离后,采用SYPRO Ruby凝胶染色剂染色,对差异条带进行液相色谱-串联质谱(LC-MS/MS)分析。结果显示,与对照组相比,FUS-DDIT3和DDIT3表达组中,CDK2与细胞骨架蛋白plectin、肌球蛋白(myosin)、波形蛋白(vimentin)的结合亲和力显著增强,这些蛋白在对照组的免疫沉淀复合物中未被检测到(n=3,文献未明确提供P值,基于图表趋势推测)。实验所用关键产品:SYPRO Ruby凝胶染色剂(Sigma-Aldrich)、LC-MS/MS检测(哥德堡大学蛋白质组学核心设施)。
4. Biomarker研究及发现成果
本研究未聚焦疾病诊断或预后Biomarker的筛选与验证,而是阐明了MLS/RCLS驱动癌蛋白FUS-DDIT3与细胞周期调控因子CDK2的相互作用机制,该分子事件可作为MLS/RCLS潜在治疗靶点的核心基础。
Biomarker定位方面,本研究未鉴定新的疾病诊断、预后或疗效预测Biomarker,而是揭示了FUS-DDIT3与CDK2的直接相互作用作为MLS/RCLS恶性转化的关键分子机制,为后续开发靶向该相互作用的治疗策略提供了理论依据。研究过程中,通过免疫荧光、免疫共沉淀、突变体实验等多种技术证实,FUS-DDIT3通过其DDIT3结构域的N端区域直接结合CDK2,且该结合不依赖亮氨酸拉链结构;进一步的功能分析显示,该相互作用不改变CDK2的表达水平、磷酸化状态及蛋白半衰期,但显著改变CDK2与细胞骨架蛋白的结合谱,提示FUS-DDIT3可能通过调控CDK2的细胞骨架相关功能参与肿瘤细胞的恶性行为。核心成果提炼为首次证实DDIT3和FUS-DDIT3可直接结合CDK2,揭示了FUS-DDIT3调控细胞周期和细胞骨架的新机制,为MLS/RCLS的分子机制研究提供了新的切入点;本研究未涉及临床样本验证,也未提供该相互作用与患者预后或治疗响应的关联数据,需进一步体内实验和临床研究证实其转化应用价值。