1. 领域背景与文献引入
文献英文标题:Cellular Notch responsiveness is defined by phosphoinositide 3-kinase-dependent signals;发表期刊:BMC Cell Biology;影响因子:未公开;研究领域:细胞信号转导(Notch与PI3K通路交叉调控)
领域共识:Notch信号通路是进化保守的细胞间通讯通路,1999年《Science》发表的综述明确其在胚胎发育、细胞命运决定中的核心作用;磷脂酰肌醇3激酶(PI3K)-蛋白激酶B(Akt)通路是调控细胞增殖、存活、分化的关键通路,2002年《Science》研究证实其在淋巴细胞激活中的必需功能。当前研究热点聚焦于不同信号通路的交叉调控机制,尤其是在免疫细胞功能、神经发育等过程中的作用,未解决的核心问题包括Notch与PI3K通路的具体调控关系、该调控是否具有细胞类型普遍性。
结合领域现状,现有研究发现p110δ缺陷小鼠与Notch2缺陷小鼠均存在边缘区B细胞发育障碍,提示两条通路可能存在功能关联,但具体调控机制尚未明确。本研究旨在解析PI3K-Akt通路对Notch反应性的调控作用及分子机制,其学术价值在于明确两条核心通路的交叉调控范式,为免疫疾病治疗、神经发育异常干预提供潜在靶点。
2. 文献综述解析
作者以“通路交叉调控的细胞特异性与分子机制”为核心逻辑,按细胞类型(T细胞、神经元、CHO细胞)和通路上下游层级(PI3K→Akt→糖原合酶激酶3β(GSK3β)→Notch)对现有研究进行分类评述。
现有研究中,Notch通路的关键结论是通过调控转录因子CBF-1激活靶基因(如Hes-1),参与细胞命运决定、免疫细胞分化及神经发育,技术方法以报告基因检测、基因敲除/过表达为主,优势是能直观反映通路活性,局限性在于不同细胞类型中Notch的功能存在差异,部分研究结论相互矛盾;PI3K-Akt通路的关键结论是通过磷酸化下游靶点调控细胞增殖、存活,技术方法包括基因靶向敲除、小分子抑制剂处理,优势是能精准调控通路活性,局限性在于通路下游靶点众多,交叉调控的具体分子机制尚未完全阐明。
通过对比现有研究的未解决问题,本研究的创新价值在于首次证明PI3K-Akt通路通过抑制GSK3β正向调控Notch反应性,且该调控机制在T细胞、神经元、CHO细胞等多种细胞类型中保守,解决了此前仅观察到表型相似但机制不明的研究空白,为通路交叉调控的普遍性提供了直接实验证据。
3. 研究思路总结与详细解析
本研究的整体框架为“假设驱动-多细胞验证-分子机制解析”,研究目标是明确PI3K-Akt通路对Notch反应性的调控作用及分子机制,核心科学问题是PI3K-Akt通路如何调控Notch信号,技术路线为:提出PI3K-Akt调控Notch的假设→在Jurkat T细胞系中验证通路调控作用→在原代人CD4+T细胞中验证生理状态下的调控→鉴定下游关键分子GSK3β→在CHO细胞和神经元中验证机制的普遍性→总结调控模型。
3.1 Jurkat T细胞中PI3K-Akt通路对Notch信号的调控
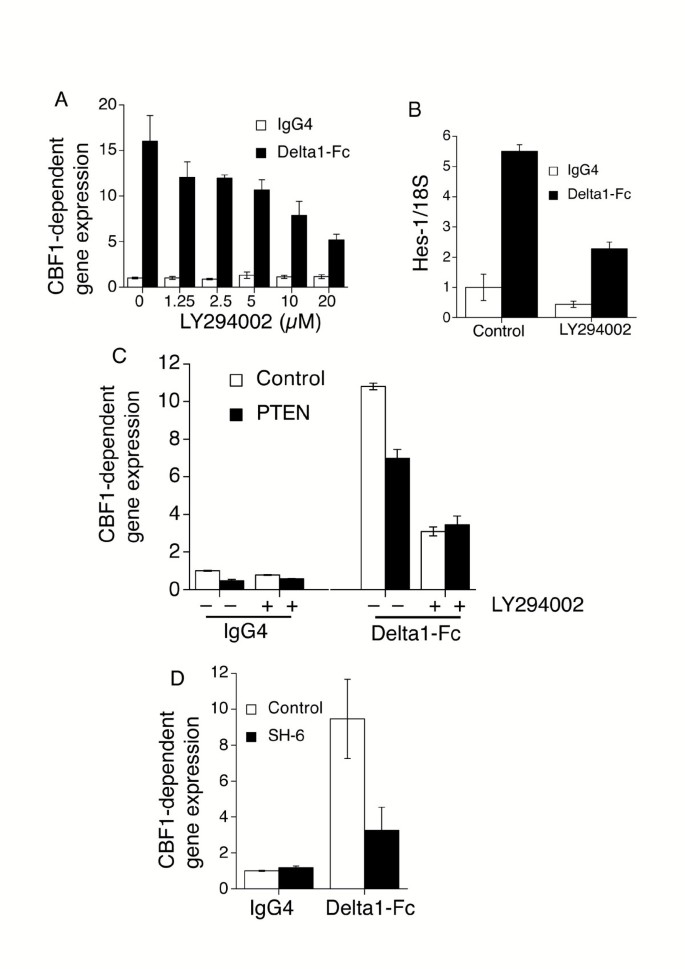
实验目的:验证PI3K-Akt通路是否为Notch信号的正向调控因子。方法细节:构建稳定表达人Notch2全长cDNA的Jurkat E6.1 T细胞系(J-N2),采用板结合重组人Delta1-Fc融合蛋白激活Notch通路,通过瞬时转染CBF1驱动的荧光素酶报告基因(p10xCBF1-luc)检测Notch活性;分别用PI3K抑制剂LY294002、Wortmannin处理细胞,或转染磷脂酶Pten(负向调控PI3K通路)、Akt抑制剂SH-6处理,设置相应对照组。结果解读:LY294002处理后,Delta1-Fc诱导的CBF1报告基因活性呈剂量依赖性降低(文献未明确具体数值,基于图表趋势推测);转染Pten后,J-N2细胞的基础及配体诱导的Notch活性均显著降低,且在LY294002处理后,Pten过表达组与对照组的报告基因活性无显著差异;Akt抑制剂SH-6处理可抑制Delta1-Fc介导的Notch信号上调,说明PI3K-Akt通路是J-N2细胞中Notch信号的关键正向调控因子。产品关联:文献未提及具体实验产品,领域常规使用荧光素酶报告基因检测试剂盒、PI3K/Akt小分子抑制剂类试剂。
3.2 原代人CD4+T细胞中PI3K-Akt通路对Notch反应性的调控
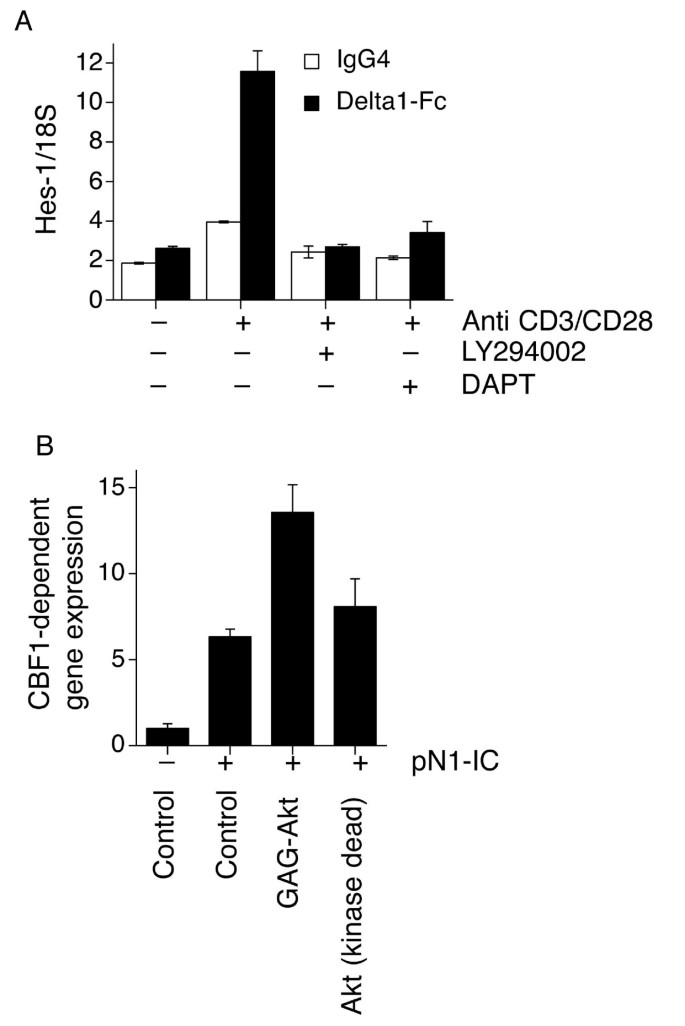
实验目的:验证PI3K-Akt通路在生理状态下的原代T细胞中对Notch反应性的调控作用。方法细节:从健康供者外周血中分离纯化CD4+T细胞,采用抗CD3/CD28抗体激活T细胞受体(TCR)通路,Delta1-Fc激活Notch通路;分别用LY294002、γ分泌酶抑制剂DAPT处理细胞,通过实时定量聚合酶链反应(qRT-PCR)检测Notch靶基因Hes-1的表达;同时采用核转染技术过表达组成型激活的Akt(gagAkt)或Notch1胞内域(N1-IC),检测CBF1报告基因活性。结果解读:在无外源性Delta1刺激时,TCR激活可上调Hes-1表达,且该效应可被LY294002和DAPT完全抑制;Delta1-Fc单独处理仅诱导Hes-1轻度上调,而与TCR激活协同处理时,Hes-1表达显著升高,且依赖PI3K通路(LY294002可抑制);过表达gagAkt可显著增强N1-IC驱动的CBF1报告基因活性,说明PI3K-Akt通路可增强原代CD4+T细胞的Notch反应性。产品关联:文献未提及具体实验产品,领域常规使用磁珠分离CD4+T细胞、qRT-PCR检测试剂盒类试剂。
3.3 GSK3β在PI3K-Akt调控Notch信号中的介导作用
实验目的:鉴定PI3K-Akt通路调控Notch信号的下游关键分子。方法细节:在原代人CD4+T细胞中,用GSK3β抑制剂氯化锂(LiCl)处理细胞,过表达N1-IC或用Delta1-Fc激活Notch通路,检测CBF1报告基因活性;在J-N2细胞(Pten缺陷,基础Akt活性高)中用LiCl处理,检测Delta1-Fc诱导的Notch活性。结果解读:LiCl处理可剂量依赖性增强原代CD4+T细胞中N1-IC诱导的Notch信号,且显著增强Delta1-Fc介导的CBF1报告基因活性;而在J-N2细胞中,LiCl处理对Delta1-Fc诱导的Notch信号无进一步增强作用,提示在Akt活性已饱和的细胞中,GSK3β已被充分抑制,无法再调控Notch信号,说明GSK3β是PI3K-Akt通路调控Notch反应性的关键下游分子。产品关联:文献未提及具体实验产品,领域常规使用GSK3β小分子抑制剂、报告基因检测系统类试剂。

3.4 CHO细胞和神经元中PI3K-Akt通路对Notch反应性的调控
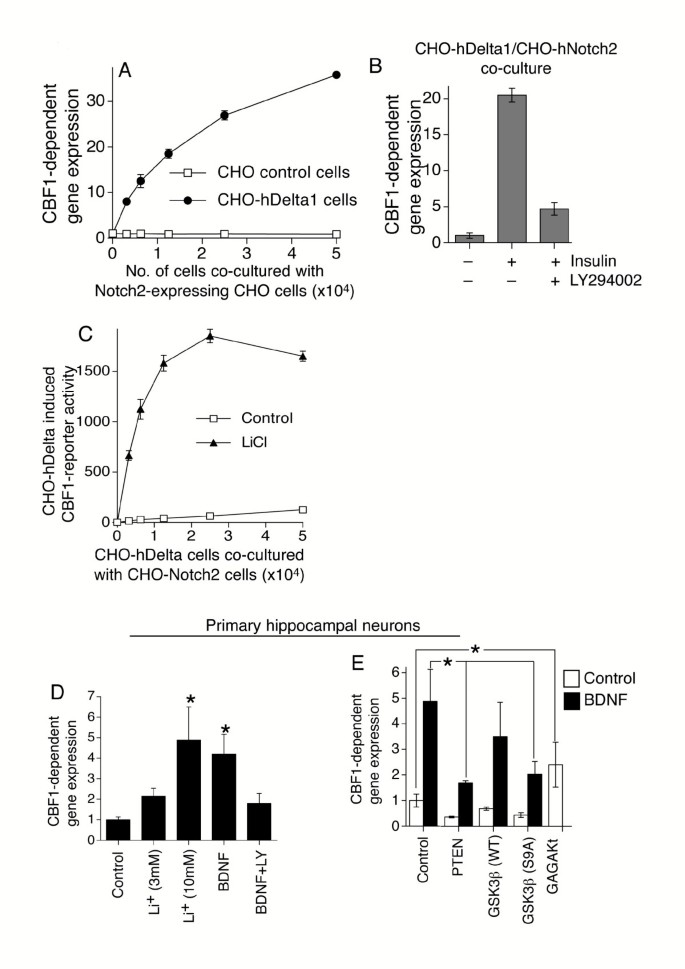
实验目的:验证PI3K-Akt-GSK3β轴调控Notch反应性的细胞类型普遍性。方法细节:采用稳定表达Notch2和CBF1报告基因的CHO-N2细胞,与表达Delta1的CHO-hDelta1细胞共培养,加入胰岛素激活PI3K通路或LY294002处理,检测报告基因活性;在原代大鼠海马神经元中,用脑源性神经营养因子(BDNF)激活PI3K通路,LiCl处理抑制GSK3β,转染Pten、gagAkt或组成型激活的GSK3β(S9A,不被Akt磷酸化抑制),检测N1-IC驱动的CBF1报告基因活性。结果解读:胰岛素处理可显著增强CHO细胞中Notch信号,LY294002可完全阻断该效应;LiCl处理可剂量依赖性增强神经元中Notch信号,BDNF处理可增强N1-IC驱动的报告基因活性,LY294002和Pten转染可阻断该效应;过表达gagAkt可增强神经元中Notch信号,而S9A GSK3β过表达可阻断BDNF对Notch信号的增强作用,说明PI3K-Akt-GSK3β轴对Notch反应性的调控在多种细胞类型中保守。产品关联:文献未提及具体实验产品,领域常规使用细胞共培养体系、神经细胞培养试剂类试剂。

4. Biomarker研究及发现成果解析
本研究中涉及的生物标志物(Biomarker)为信号通路调控分子GSK3β,其作为PI3K-Akt通路调控Notch反应性的关键节点,具有重要的功能关联与临床潜在价值。
Biomarker定位:类型为细胞内信号调控激酶(糖原合酶激酶3β,GSK3β),筛选/验证逻辑为:基于PI3K-Akt通路对GSK3β的磷酸化抑制作用,通过小分子抑制剂处理、基因过表达/敲低实验,在T细胞、神经元、CHO细胞中验证其对Notch信号的调控作用,形成“通路激活→GSK3β抑制→Notch信号增强”的完整逻辑链条。
研究过程详述:GSK3β的来源为细胞内固有激酶,验证方法包括荧光素酶报告基因检测、基因转染调控表达;特异性表现为仅在Akt活性未饱和的细胞(如原代T细胞)中,抑制GSK3β可增强Notch信号,而在Akt活性高的Jurkat细胞中无效应;敏感性表现为低浓度LiCl处理即可显著增强Notch信号(文献未明确具体敏感性数值,基于图表趋势推测)。
核心成果提炼:GSK3β的功能关联为负向调控Notch信号,PI3K-Akt通路通过磷酸化GSK3β使其失活,从而解除对Notch信号的抑制;创新性在于首次证明PI3K-Akt-GSK3β轴对Notch反应性的保守调控作用,为通路交叉调控提供了关键分子节点;统计学结果包括神经元实验中,gagAkt过表达组与对照组相比,报告基因活性显著升高(n=3,P<0.05,Mann Whitney U检验),其余实验数据以均值±标准差表示,为三次独立重复实验的代表性结果。