1. 领域背景与文献引入
文献英文标题:FBXL17/spastin axis as a novel therapeutic target of hereditary spastic paraplegia;发表期刊:Cell & Bioscience;影响因子:未公开;研究领域:遗传性痉挛性截瘫(HSP)的分子机制与治疗靶点研究
遗传性痉挛性截瘫是一组以进行性下肢痉挛和无力为核心特征的神经退行性疾病,全球患病率约为1/10000至1/20000。SPAST基因(编码spastin蛋白)突变是常染色体显性HSP最常见的病因,占比超过40%。领域共识:spastin蛋白在微管切割、轴突运输和内质网动态调控中发挥关键作用,其功能缺失会导致轴突肿胀、细胞器积累和神经元功能障碍。当前研究热点集中在spastin的转录调控、突变体的ATP酶活性缺陷机制,但spastin蛋白的翻译后修饰调控(尤其是泛素化修饰)尚未明确,这是领域未解决的核心问题。现有治疗手段仅能通过康复训练和肌肉松弛剂缓解症状,缺乏针对病因的精准治疗策略,因此解析spastin的翻译后调控机制对开发HSP新型治疗靶点至关重要。本研究针对spastin-M1的泛素化调控机制空白,旨在揭示FBXL17-spastin轴在HSP致病中的作用,为HSP提供新的治疗方向。
2. 文献综述解析
作者对领域内现有研究的分类维度主要为spastin的调控机制(转录调控、翻译后修饰)和HSP的致病机制(功能缺失、蛋白积累)。现有研究表明,spastin的转录调控受NRF-1和Sox11等转录因子调控,翻译后修饰方面仅发现HIPK2介导的磷酸化,泛素化调控机制完全未知;spastin突变体的致病机制主要集中在ATP酶活性丧失和微管切割功能缺陷,但部分突变体(如Y52C)的致病机制未被解析。现有研究的技术方法优势在于建立了多种HSP细胞和动物模型,可有效模拟疾病表型,但局限性在于缺乏对spastin蛋白稳定性调控的深入研究,尤其是泛素-蛋白酶体系统在其中的作用,且未建立spastin调控通路与HSP治疗的关联。
本研究的创新价值在于首次发现FBXL17作为E3泛素连接酶通过SCF复合物调控spastin-M1的核内泛素化降解,填补了spastin翻译后修饰调控的空白;同时揭示了FBXL17-spastin轴在HSP致病中的作用,为HSP提供了新的治疗靶点,突破了现有研究仅关注spastin功能突变的局限,拓展了HSP致病机制的研究维度。
3. 研究思路总结与详细解析
本研究的整体框架为:以“解析spastin-M1的泛素化调控机制及其在HSP中的致病作用”为研究目标,核心科学问题是“FBXL17如何调控spastin-M1的蛋白稳定性”,技术路线遵循“蛋白芯片筛选互作蛋白→验证相互作用与调控机制→解析翻译后修饰的 Crosstalk→验证调控通路的治疗潜力→突变体致病机制解析”的闭环逻辑。
3.1 spastin-M1互作E3泛素连接酶的筛选与验证
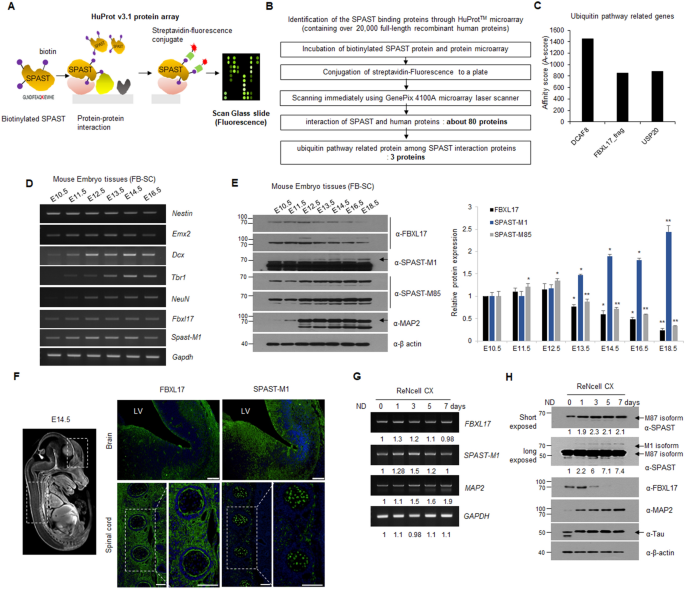
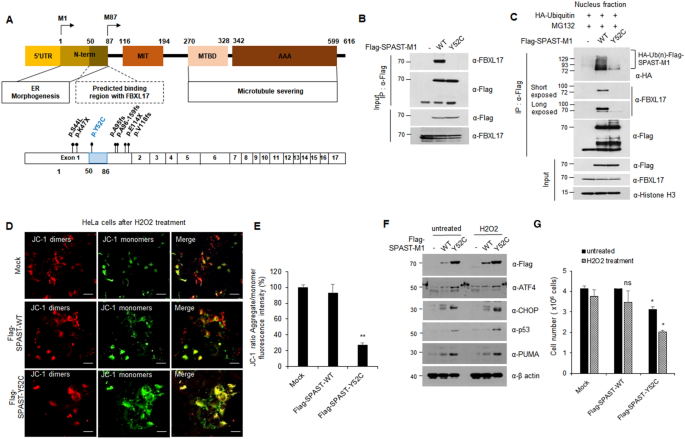
实验目的是筛选并验证调控spastin-M1的E3泛素连接酶。方法细节为使用包含20000余种人类重组蛋白的HuProt™蛋白芯片,以生物素化的His-spastin-M1为诱饵筛选互作蛋白,通过免疫共沉淀(Co-IP)和免疫印迹(WB)验证互作;同时检测小鼠胚胎脑和脊髓组织、ReNcell CX神经分化过程中spastin-M1与FBXL17的表达相关性。结果解读显示,蛋白芯片筛选出FBXL17等4个泛素通路相关蛋白,Co-IP验证spastin-M1仅与FBXL17直接互作;在小鼠胚胎发育阶段(E10.5-E18.5)和ReNcell CX神经分化过程中,spastin-M1蛋白表达与FBXL17呈负相关(n=3,P<0.05)。实验所用关键产品:HuProt™蛋白芯片(CDI Labs)、GenePix4100A微阵列激光扫描仪(Molecular Devices)。
3.2 spastin-M1与FBXL17的互作结构域解析
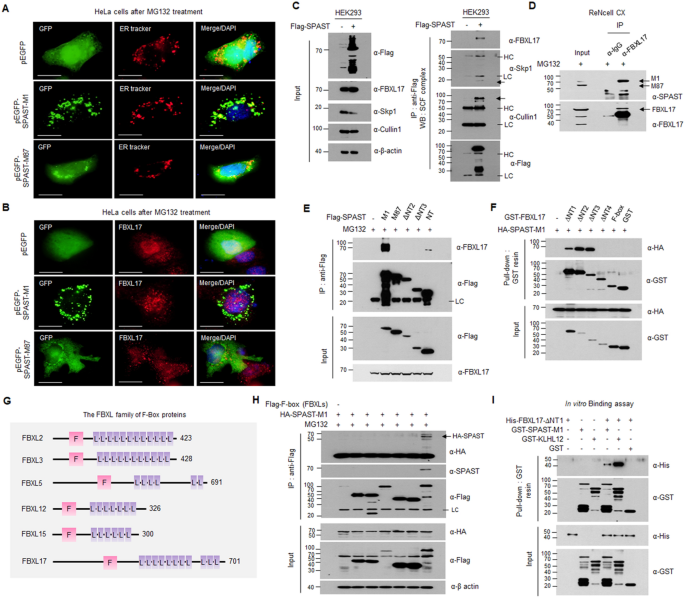
实验目的是明确spastin-M1与FBXL17互作的具体结构域。方法细节为构建spastin-M1的N端截短突变体和FBXL17的截短突变体,通过Co-IP、GST pull-down和体外结合实验验证互作结构域;同时检测spastin-M1和FBXL17的亚细胞定位。结果解读显示,spastin-M1通过N端的BTB结构域(50-86 aa)与FBXL17的亮氨酸重复序列(LRR)结构域(471-571 aa)互作,spastin-M87因缺乏BTB结构域无法与FBXL17互作;spastin-M1定位于内质网和细胞核,FBXL17主要定位于细胞核,二者在细胞核中共定位。实验所用关键产品:CRISPR/Cas9质粒(Santa Cruz)、免疫荧光显微镜(IX71,Olympus)。
3.3 SCF^{FBXL17}复合物介导spastin-M1的核内泛素化降解
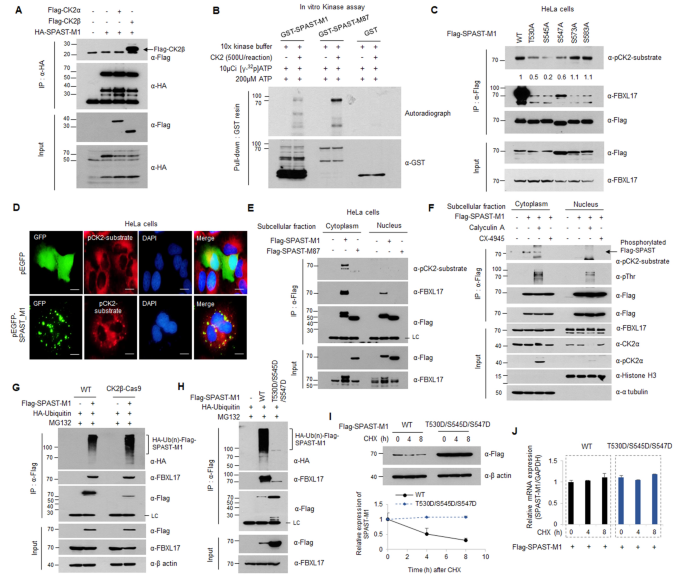
实验目的是验证SCF^{FBXL17}复合物对spastin-M1的泛素化降解作用及亚细胞定位特异性。方法细节为在HEK293T和ReNcell CX细胞中过表达或敲低FBXL17,通过WB检测spastin-M1的蛋白水平;使用环己酰亚胺(CHX)检测spastin-M1的蛋白稳定性;分离细胞核和细胞质组分,通过体内泛素化实验检测spastin-M1的泛素化水平。结果解读显示,FBXL17剂量依赖性下调spastin-M1蛋白水平(n=3,P<0.01),MG132处理可抑制该降解,且不影响spastin-M1的mRNA水平;spastin-M1的半衰期在FBXL17过表达时从>4h缩短至约3h;spastin-M1的多聚泛素化仅发生在细胞核组分中,K554是主要的泛素化位点。实验所用关键产品:NE-PER核质分离试剂盒(Thermo Fisher Scientific)、MG132(Sigma-Aldrich)。

3.4 CK2介导的spastin-M1磷酸化调控其泛素化
实验目的是解析磷酸化与泛素化在spastin-M1调控中的 Crosstalk。方法细节为通过Co-IP验证spastin-M1与CK2的互作,体外激酶实验检测CK2对spastin-M1的磷酸化;构建spastin-M1的磷酸化位点突变体,通过WB和泛素化实验检测磷酸化对泛素化的影响;使用CK2抑制剂CX4945和磷酸酶抑制剂calyculin A验证磷酸化的调控作用。结果解读显示,spastin-M1与CK2β互作,CK2可在细胞质中磷酸化spastin-M1的T530、S545和S547位点;磷酸化的spastin-M1可逃避SCF^{FBXL17}复合物的泛素化降解,蛋白稳定性显著提高(n=3,P<0.01)。实验所用关键产品:CK2激酶(New England Biolabs)、CX4945(Sigma-Aldrich)。
3.5 抑制SCF^{FBXL17}复合物对HSP细胞模型的治疗潜力验证
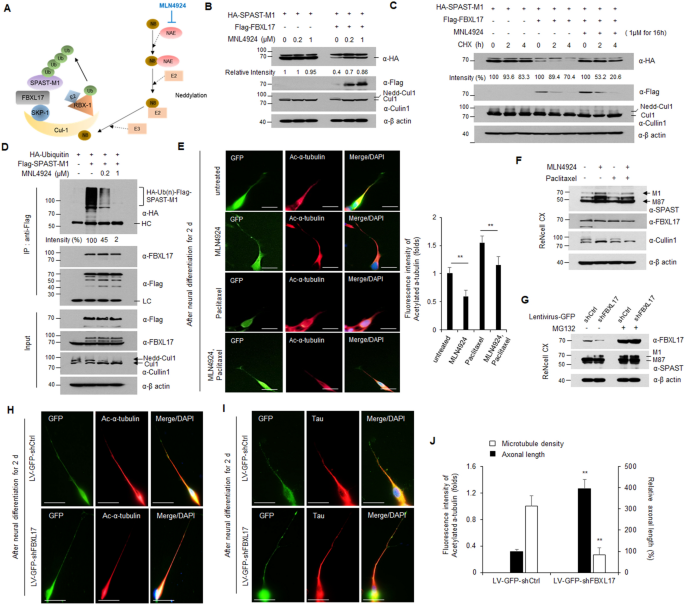
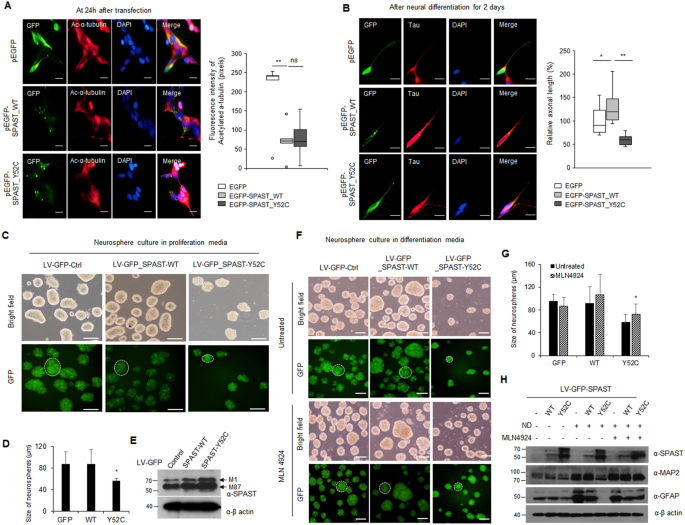
实验目的是验证抑制FBXL17对spastin-M1蛋白水平和轴突生长的影响。方法细节为使用MLN4924(Neddylation抑制剂,抑制SCF复合物活性)处理HEK293T和ReNcell CX细胞,通过WB检测spastin-M1的蛋白水平;使用紫杉醇构建轴突损伤模型,通过免疫荧光检测轴突长度;构建FBXL17的shRNA慢病毒,敲低ReNcell CX细胞中的FBXL17,检测轴突生长情况。结果解读显示,MLN4924处理可剂量依赖性提高spastin-M1蛋白水平(n=3,P<0.01),并恢复紫杉醇诱导的轴突缩短和肿胀;FBXL17敲低可显著提高spastin-M1蛋白水平,促进轴突延伸(轴突长度增加约2.1倍,n=3,P<0.01)。实验所用关键产品:MLN4924(Selleck)、慢病毒载体(OriGene)。
3.6 spastin Y52C突变体的致病机制与治疗干预
实验目的是解析spastin Y52C突变体的致病机制及MLN4924的治疗效果。方法细节为构建spastin Y52C突变体,通过Co-IP验证其与FBXL17的互作;在ReNcell CX细胞中过表达Y52C突变体,通过免疫荧光检测轴突生长、3D神经球实验检测增殖和分化能力;使用MLN4924处理突变体过表达的细胞,检测表型恢复情况。结果解读显示,Y52C突变体因BTB结构域破坏无法与FBXL17互作,逃避泛素化降解,蛋白积累但功能丧失;过表达Y52C突变体导致轴突长度缩短约35%(n=3,P<0.01),3D神经球直径减小约40%(n=3,P<0.01),分化能力下降;MLN4924处理可恢复突变体的轴突生长和分化能力。实验所用关键产品:3D细胞培养板(Corning)、Tau抗体(Cell Signaling Technology)。
4. Biomarker研究及发现成果解析
Biomarker定位
本研究中涉及的Biomarker主要是spastin-M1蛋白及其Y52C突变体。筛选逻辑基于SPAST基因在HSP中的高突变率,通过蛋白芯片筛选其调控因子,进而解析突变体的致病机制;验证逻辑遵循“细胞系验证→动物胚胎组织验证→临床突变体验证”的完整链条,确保结果的可靠性。
研究过程详述
spastin-M1来源于人类神经元细胞,通过免疫印迹、免疫荧光检测其表达水平;Y52C突变体来源于日本HSP患者,通过基因克隆构建突变体表达载体。验证方法包括免疫共沉淀、体内泛素化实验、3D神经球实验等,其中spastin-M1与FBXL17的互作具有特异性,Y52C突变体的特异性表现为无法与FBXL17结合,其在细胞中的积累与HSP的轴突损伤表型直接相关。原文未提供ROC曲线、敏感性和特异性的具体数据。
核心成果提炼
本研究核心成果包括:首次揭示spastin-M1的蛋白稳定性由FBXL17-SCF复合物和CK2共同调控,其中FBXL17介导核内泛素化降解,CK2介导的细胞质磷酸化可抑制该过程;Y52C突变体通过逃避FBXL17介导的降解导致功能丧失和蛋白异常积累,是HSP的致病机制之一;首次发现FBXL17作为HSP的潜在治疗靶点,抑制FBXL17可恢复spastin-M1的蛋白水平和功能,为HSP的精准治疗提供了新方向。相关统计学结果包括FBXL17过表达导致spastin-M1半衰期缩短(P<0.01),Y52C突变体导致轴突长度缩短(P<0.01),MLN4924处理可显著恢复轴突生长(P<0.01)。