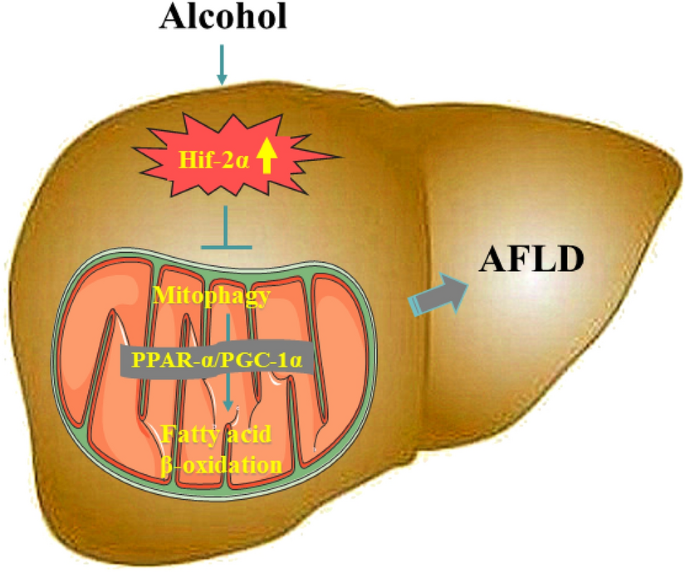
1. 领域背景与文献引入
文献英文标题:Hif-2α regulates lipid metabolism in alcoholic fatty liver disease through mitophagy;发表期刊:Cell & Bioscience;影响因子:未公开;研究领域:酒精性脂肪肝疾病的脂质代谢调控机制
酒精性肝病(ALD)是全球范围内严重的公共健康问题,酒精性脂肪肝疾病(AFLD)作为ALD的初始阶段,以肝细胞脂质积累、轻度炎症和损伤为核心特征,若未及时干预可进展为酒精性肝炎、肝硬化甚至肝细胞癌。领域共识:AFLD的发病机制涉及乙醇诱导的线粒体功能异常、氧化应激及脂质代谢紊乱,其中脂肪酸β-氧化缺陷是导致肝细胞脂质积累的关键因素之一。当前研究热点聚焦于脂质代谢调控通路、线粒体自噬的保护作用及潜在治疗靶点,但Hif-2α在AFLD中的具体调控机制,尤其是其与线粒体自噬、脂肪酸β-氧化的关联尚未明确,存在核心机制空白。本研究针对这一空白,旨在揭示Hif-2α在AFLD中的功能及调控通路,为AFLD的防治提供新的理论依据和治疗靶点。
2. 文献综述解析
作者从AFLD的核心病理机制、脂质代谢调控的关键通路、Hif家族与脂质代谢的关联、线粒体自噬在肝病中的作用四个维度对现有研究进行分类评述。
现有研究的关键结论显示,AFLD的核心病理特征是肝细胞脂质积累,脂肪酸β-氧化是肝细胞脂质清除的主要途径,其功能缺陷直接导致脂质积累;Hif-2α作为核受体家族成员,参与调控肝脏脂肪酸β-氧化的关键酶,在脂质代谢中发挥重要作用;线粒体自噬作为选择性自噬的一种,可清除受损线粒体,维持肝细胞脂质稳态,其功能异常与脂肪肝疾病进展相关。技术方法方面,现有研究多采用经典的动物模型(如慢性酒精喂养模型)和细胞模型结合分子生物学技术(如qRT-PCR、蛋白质免疫印迹)验证机制,具有较好的重复性和可靠性。但现有研究存在明显局限性,未明确Hif-2α、线粒体自噬、脂肪酸β-氧化三者在AFLD中的直接调控通路,缺乏系统性的机制验证,且未针对这一通路提出针对性的治疗策略。
本研究的创新价值在于首次揭示了Hif-2α-BNIP3依赖的线粒体自噬调控通路,直接连接乙醇诱导的脂质积累与脂肪酸β-氧化缺陷,明确了Hif-2α在AFLD中的促进展作用,为AFLD的防治提供了新的潜在治疗靶点,弥补了现有研究的机制空白。
3. 研究思路总结与详细解析
本研究的研究目标是明确Hif-2α在AFLD中的功能及调控机制,核心科学问题是Hif-2α如何通过线粒体自噬调控脂肪酸β-氧化进而影响AFLD的进展,技术路线遵循“体内外表达验证→功能抑制验证→机制通路探究→模型总结”的闭环逻辑,通过多层次实验验证Hif-2α的调控作用及分子机制。
3.1 体内外Hif-2α表达水平验证
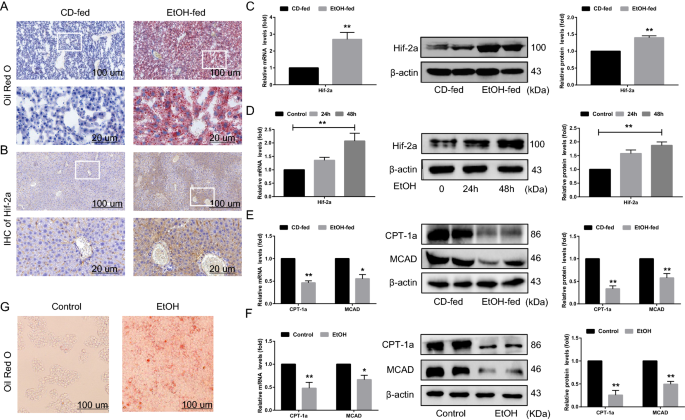
本环节的实验目的是确认Hif-2α在AFLD模型中的表达变化,明确其与AFLD的关联。实验方法采用NIAAA推荐的慢性加 binge 喂养方法构建雄性C57B6/L小鼠AFLD模型,给予Lieber-De Carli液体饮食含5%乙醇10天,同时设置对照组;细胞实验采用小鼠肝细胞系AML-12,用100mM乙醇处理48h。通过油红O染色观察脂质积累,免疫组化(IHC)、qRT-PCR、蛋白质免疫印迹检测Hif-2α及脂肪酸β-氧化关键酶CPT-1α、MCAD的表达水平。实验结果显示,与对照组相比,乙醇喂养小鼠肝脏脂质滴积累显著增加(n=6,P<0.05),肝损伤指标血清AST、ALT、TG、T-CHO水平升高;免疫组化、qRT-PCR及蛋白质免疫印迹结果一致显示,小鼠肝脏和AML-12细胞中Hif-2α表达显著上调(n=6/3,P<0.05),同时CPT-1α和MCAD的表达显著下调(n=6/3,P<0.05),提示Hif-2α的上调与AFLD的脂质积累相关。实验所用关键产品:TRIzol Reagent(Invitrogen)、SYBR® Prime Script™ RT–PCR Kit(TAKARA)、Hif-2α抗体(Abcam)、CPT-1α和MCAD抗体(Proteintech)、油红O染色试剂。
3.2 Hif-2α对脂肪酸β-氧化的调控作用验证
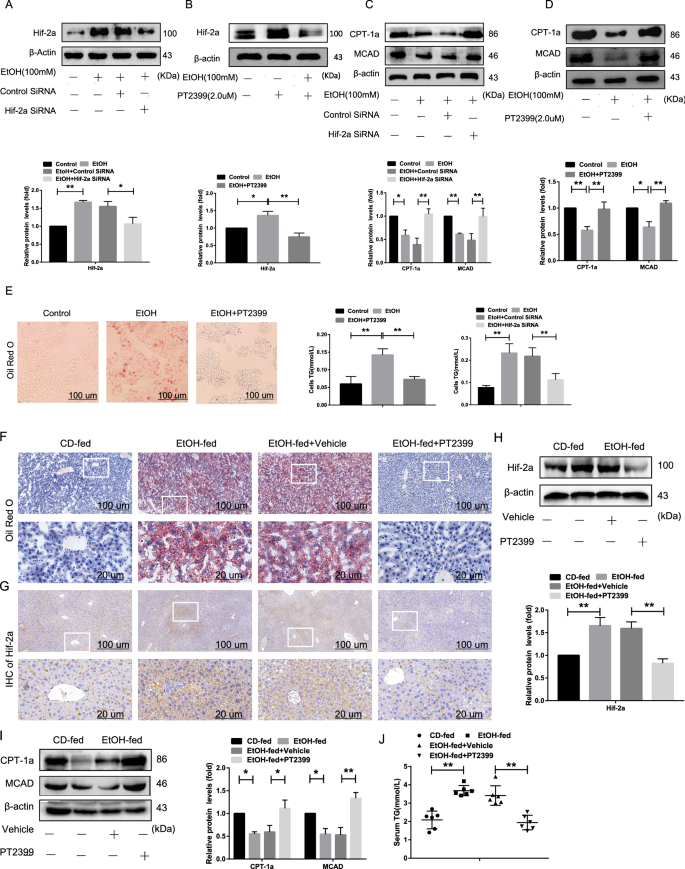
本环节的实验目的是明确Hif-2α与脂肪酸β-氧化的直接关联,验证抑制Hif-2α对AFLD表型的改善作用。实验方法在细胞层面,将AML-12细胞转染Hif-2α siRNA或用Hif-2α特异性抑制剂PT2399(2.0μM)处理48h;动物层面,对乙醇喂养小鼠灌胃PT2399(10mg/kg)7天。通过蛋白质免疫印迹检测CPT-1α、MCAD的表达,油红O染色、生化检测观察脂质积累及肝损伤指标。实验结果显示,与仅乙醇处理组相比,Hif-2α siRNA转染或PT2399处理组的CPT-1α和MCAD表达显著升高(n=3/6,P<0.05),细胞内TG水平和脂质滴数量显著降低(n=3,P<0.05);小鼠肝脏的脂质积累和肝损伤程度显著减轻,血清ALT、AST、TG、T-CHO水平及肝体比显著下降(n=6,P<0.05),提示抑制Hif-2α可通过上调脂肪酸β-氧化缓解AFLD的脂质积累和肝损伤。实验所用关键产品:Hif-2α siRNA(Gene Pharma)、PT2399、LipofectamineTM2000(Invitrogen)、生化检测试剂盒(Jiancheng)。
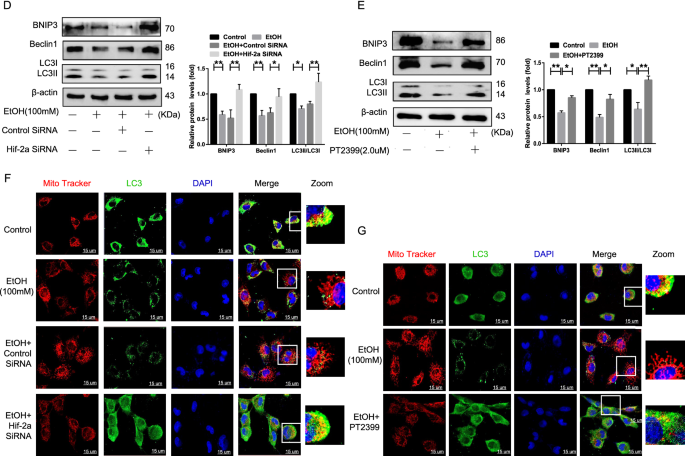
3.3 Hif-2α对BNIP3依赖的线粒体自噬的调控
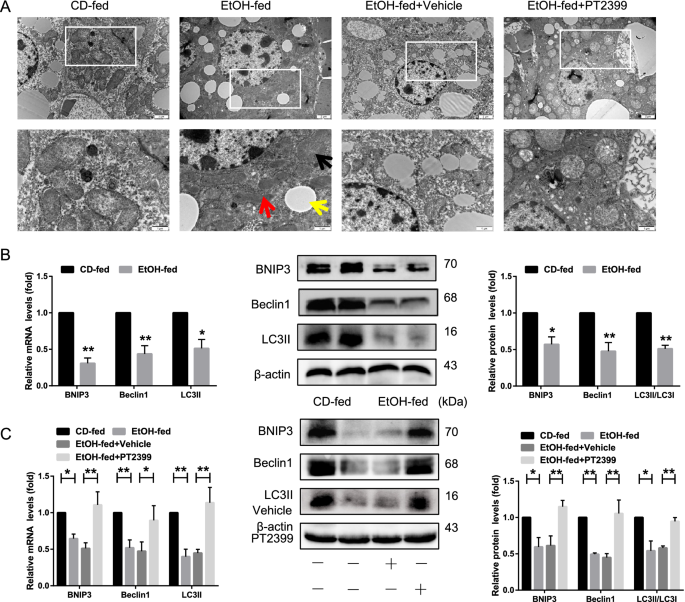
本环节的实验目的是探究Hif-2α是否通过调控线粒体自噬影响脂质代谢。实验方法采用透射电镜观察小鼠肝脏线粒体形态及自噬体数量,qRT-PCR、蛋白质免疫印迹检测线粒体自噬标志物BNIP3、Beclin1、LC3II的表达,免疫荧光染色观察LC3与线粒体的共定位情况;同时通过共转染Hif-2α siRNA与BNIP3 shRNA验证BNIP3的介导作用。实验结果显示,乙醇喂养小鼠的线粒体形态异常(肿胀、裂变减少),自噬体数量减少,线粒体自噬标志物BNIP3、Beclin1、LC3II的表达显著下调(n=6,P<0.05);抑制Hif-2α后,线粒体自噬标志物的表达显著上调(n=6/3,P<0.05),LC3与线粒体的共定位增加(n=3,P<0.05);而共转染BNIP3 shRNA则逆转了Hif-2α沉默对线粒体自噬的激活作用,提示Hif-2α通过抑制BNIP3依赖的线粒体自噬参与AFLD的进展。实验所用关键产品:Mito Tracker Red(Invitrogen)、BNIP3 shRNA(Gene Pharma)、免疫荧光抗体、透射电镜相关试剂。
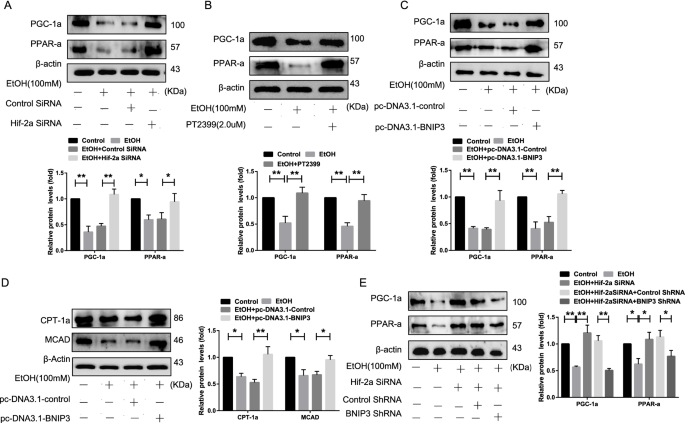
3.4 Hif-2α通过BNIP3-线粒体自噬调控PPAR-α/PGC-1α通路
本环节的实验目的是明确Hif-2α调控脂肪酸β-氧化的下游信号通路。实验方法通过转染pc-DNA3.1-BNIP3质粒过表达BNIP3,或共转染Hif-2α siRNA与BNIP3 shRNA,采用蛋白质免疫印迹检测PPAR-α、PGC-1α及脂肪酸β-氧化关键酶的表达水平。实验结果显示,过表达BNIP3可显著上调PPAR-α、PGC-1α的表达(n=3,P<0.05),同时CPT-1α和MCAD的表达也显著升高;而共转染BNIP3 shRNA则逆转了Hif-2α沉默对PPAR-α/PGC-1α通路的激活作用,提示Hif-2α通过抑制BNIP3依赖的线粒体自噬,进而下调PPAR-α/PGC-1α通路,最终抑制脂肪酸β-氧化。实验所用关键产品:pc-DNA3.1-BNIP3质粒(Gene Pharma)、PPAR-α抗体(Cell Signaling Technology)、PGC-1α抗体(Proteintech)。
4. Biomarker研究及发现成果解析
本研究中涉及的Biomarker为Hif-2α,属于核受体蛋白类Biomarker,其筛选与验证逻辑遵循“体内外模型筛选表达异常→功能验证表型变化→机制通路验证调控作用”的完整链条。
Biomarker的来源为AFLD模型小鼠的肝脏组织及乙醇处理的AML-12肝细胞,验证方法包括免疫组化、qRT-PCR、蛋白质免疫印迹,通过多技术交叉验证Hif-2α的表达变化及定位。特异性方面,乙醇处理后Hif-2α在细胞核中的表达显著升高(n=3,P<0.05),而细胞质中表达降低,提示其核转录活性增强;敏感性方面,抑制Hif-2α的表达可显著改善AFLD的脂质积累和肝损伤表型,各指标差异具有统计学意义(P<0.05),但原文未提供ROC曲线等特异性与敏感性的量化数据。核心成果方面,Hif-2α作为AFLD的促进展Biomarker,其功能是通过抑制BNIP3依赖的线粒体自噬,下调PPAR-α/PGC-1α信号通路,进而抑制脂肪酸β-氧化,最终促进肝脂肪变性和AFLD的进展;创新性在于首次在AFLD中发现Hif-2α与BNIP3依赖的线粒体自噬、PPAR-α/PGC-1α通路的直接调控关系,明确了Hif-2α的促进展作用,为AFLD的防治提供了新的潜在治疗靶点。统计学结果显示,各关键实验指标的组间差异均具有统计学意义(P<0.05),样本量符合实验要求(动物实验n=6,细胞实验n=3)。