1. 领域背景与文献引入
文献英文标题:Degradation of the LDL receptors by PCSK9 is not mediated by a secreted protein acted upon by PCSK9 extracellularly;发表期刊:BMC Cell Biology;影响因子:未公开;研究领域:脂蛋白代谢与细胞生物学(PCSK9调控低密度脂蛋白受体机制)
领域共识:胆固醇稳态调控是心血管疾病研究的核心方向,低密度脂蛋白受体(LDLR)是清除血液中低密度脂蛋白胆固醇(LDL-C)的关键分子,其表达水平直接影响血胆固醇稳态。2003年,前蛋白转化酶枯草溶菌素9(PCSK9)被发现属于枯草酶家族,其合成后经自催化切割分泌,可通过转录后调控降低LDLR水平,进而升高血LDL-C;PCSK9的功能获得性突变与家族性高胆固醇血症相关,功能缺失性突变则导致低胆固醇血症,提示其是降脂药物的潜在靶点。现有研究已证实PCSK9可分泌至细胞外,作用于未转染细胞的LDLR,但尚未明确其降解LDLR的具体机制:是直接作用于LDLR,还是通过激活胞外其他分泌蛋白间接发挥作用?LDLR的降解发生在细胞表面还是胞内?是否依赖经典的网格蛋白介导的内吞途径?这些核心问题的解决对深入理解胆固醇代谢调控网络、开发更精准的降脂策略具有重要意义。本文针对上述空白,通过一系列细胞与分子生物学实验,系统解析了PCSK9降解LDLR的关键机制。
2. 文献综述解析
本文综述部分围绕PCSK9的基本特性、与LDLR的调控关系及现有机制争议展开,作者按“PCSK9基础生物学-PCSK9对LDLR的调控效应-机制争议点”的维度梳理领域研究。
现有研究已明确PCSK9作为分泌型蛋白酶,其合成、自催化切割及分泌过程,以及其突变与血脂异常疾病的关联;同时证实PCSK9可通过转录后机制降低LDLR蛋白水平,且该效应具有细胞特异性(仅在肝、肾细胞中发挥作用,成纤维细胞中无效应)。技术方法上,现有研究多采用细胞转染、动物模型等手段,能直观观察PCSK9对LDLR的调控效应,但存在机制解析不深入的局限性:尚未明确PCSK9是否依赖胞外其他蛋白发挥作用,也未清晰界定LDLR降解的亚细胞位置,部分研究结果存在矛盾(如ARH敲除小鼠实验提示LDLR内吞可能非必需,但其他研究认为内吞是必要步骤)。本文的创新价值在于,通过亲和层析、凝胶过滤等技术直接证明PCSK9是条件培养基中唯一降解LDLR的因子,排除了胞外中间蛋白的参与;通过截短LDLR、膜组分实验明确降解发生在胞内而非细胞表面;通过高渗介质、诺考达唑等实验揭示降解不依赖网格蛋白介导的内吞,需要微管运输至酸性胞内区室,填补了领域内机制解析的关键空白。
3. 研究思路总结与详细解析
本文的研究目标是明确PCSK9降解LDLR的分子机制,核心科学问题包括PCSK9是否直接作用于LDLR、LDLR降解的亚细胞位置及所需的胞内途径,技术路线遵循“假设验证-机制解析-途径确认”的闭环逻辑:构建不同标签的PCSK9表达载体→制备含PCSK9的条件培养基→验证是否依赖胞外其他蛋白→确定降解位置→解析胞内降解途径。
3.1 条件培养基制备与PCSK9亲和层析验证
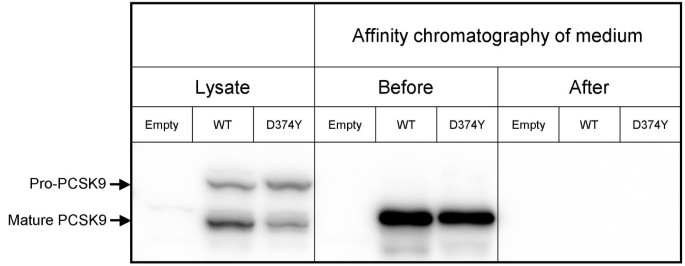
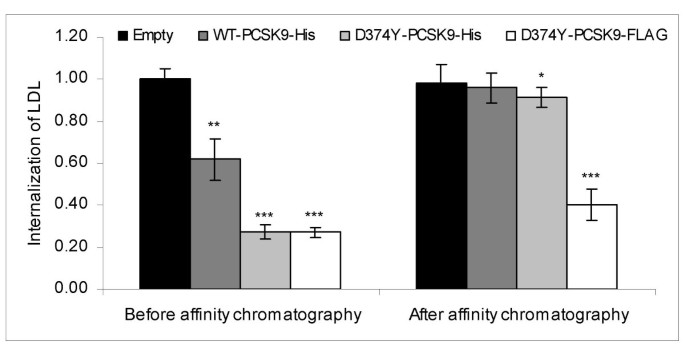
实验目的:验证PCSK9是否是条件培养基中唯一介导LDLR降解的因子,排除胞外其他分泌蛋白的参与。方法细节:将野生型(WT)PCSK9-His、功能获得性突变体D374Y-PCSK9-His及空质粒分别瞬时转染至HepG2细胞,24小时后更换无血清培养基,继续培养24小时收集条件培养基;采用镍螯合树脂亲和层析去除条件培养基中的PCSK9-His,通过免疫印迹(Western blot)检测PCSK9的去除效率,利用流式细胞术检测处理后的条件培养基对未转染HepG2细胞LDL内吞的影响。结果解读:免疫印迹结果显示,亲和层析可完全去除条件培养基中的PCSK9-His(图1);流式细胞术结果显示,含WT-PCSK9-His的条件培养基可抑制38%±10%的LDL内吞(n=4,P<0.01),含D374Y-PCSK9-His的条件培养基可抑制73%±3%的LDL内吞(n=4,P<0.0001);去除PCSK9后,两组的LDL内吞均显著恢复(P<0.05、P<0.001),而不结合镍树脂的D374Y-PCSK9-FLAG对照组内吞抑制无明显恢复(图2),提示PCSK9是条件培养基中唯一降解LDLR的因子。产品关联:文献未提及具体实验产品,领域常规使用转染试剂、免疫印迹抗体、流式细胞仪等。
3.2 凝胶过滤验证PCSK9的直接作用
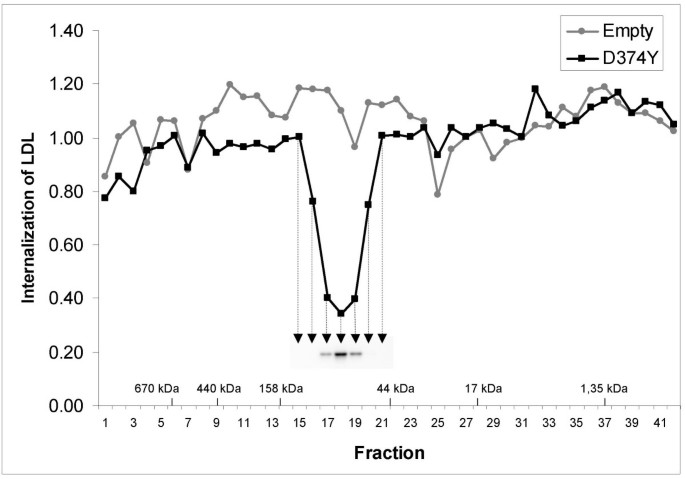
实验目的:进一步证实PCSK9自身介导LDLR降解,而非通过激活胞外其他蛋白。方法细节:将含D374Y-PCSK9-FLAG的条件培养基浓缩后,通过Superdex 200凝胶过滤层析分离为不同组分,收集各组分后孵育HepG2细胞,采用流式细胞术检测LDL内吞情况,同时通过免疫印迹检测各组分中PCSK9的含量。结果解读:组分18对LDL内吞的抑制作用最强,且该组分中PCSK9的含量最高,组分的分子流体力学体积对应约80kDa,与成熟PCSK9-前体复合物的分子量(约75kDa)接近(图3),说明LDL内吞抑制能力与PCSK9含量呈正相关,直接证明PCSK9自身是降解LDLR的效应分子,无需胞外中间蛋白参与。产品关联:文献未提及具体实验产品,领域常规使用凝胶过滤层析柱、免疫印迹抗体等。
3.3 截短LDLR与膜组分实验验证降解位置
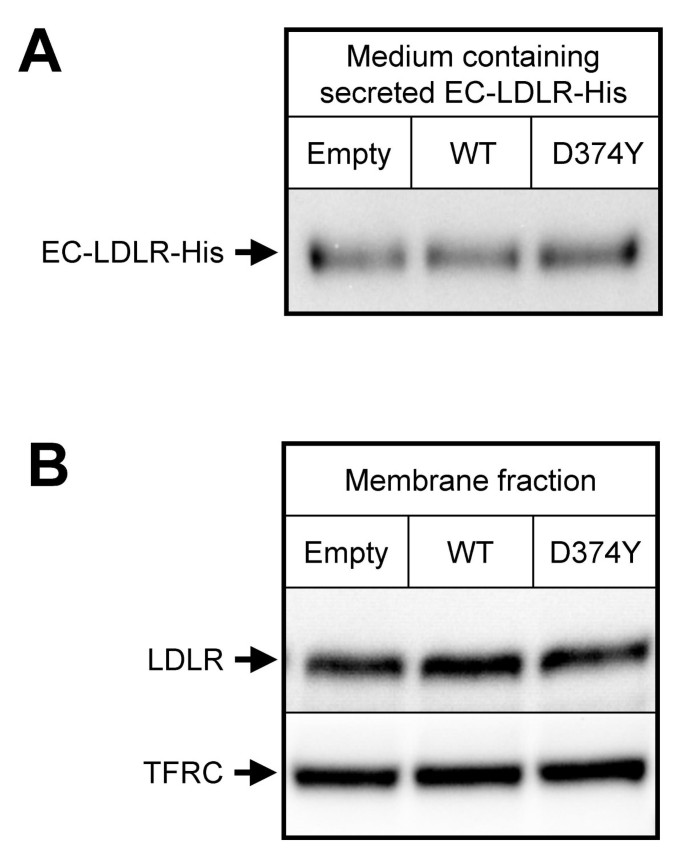
实验目的:明确PCSK9介导的LDLR降解是否发生在细胞表面。方法细节:构建缺乏胞质结构域和跨膜区的截短型LDLR(EC-LDLR-His),该蛋白可分泌至细胞外培养基;将浓缩的EC-LDLR-His与含PCSK9的条件培养基共孵育3小时;同时制备HepG2细胞膜组分,与含PCSK9的条件培养基共孵育3小时,通过免疫印迹检测LDLR的含量变化。结果解读:免疫印迹结果显示,无论是分泌型截短LDLR还是细胞膜组分中的LDLR,与PCSK9共孵育后含量均无明显降低(图4),说明PCSK9无法在细胞表面降解LDLR,需要完整细胞的胞内区室才能发挥降解效应。产品关联:文献未提及具体实验产品,领域常规使用质粒构建试剂盒、细胞膜分离试剂盒、免疫印迹抗体等。
3.4 高渗介质实验验证内吞途径非依赖性
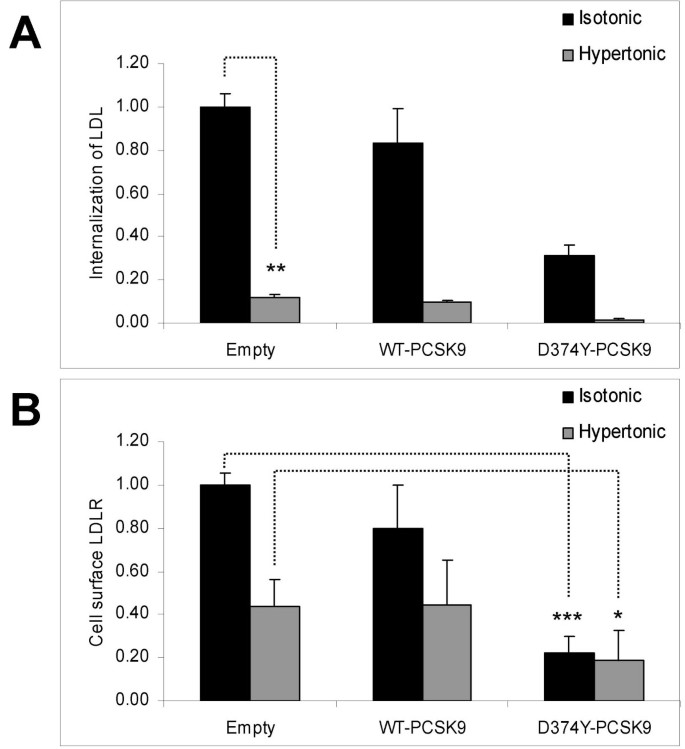
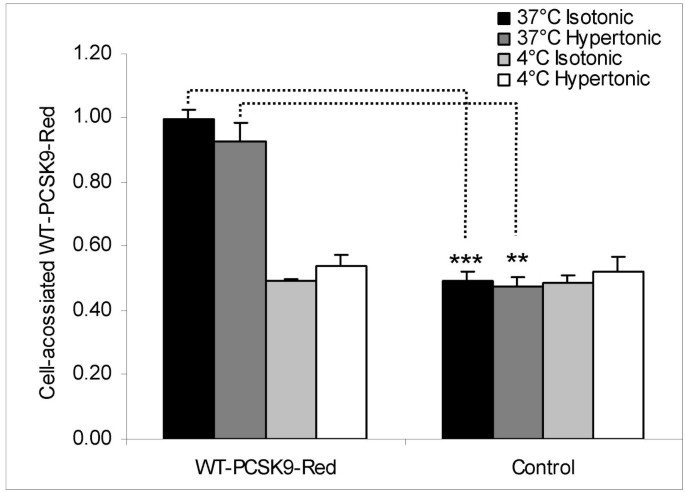
实验目的:验证PCSK9介导的LDLR降解是否依赖网格蛋白包被小窝介导的内吞途径。方法细节:在含PCSK9的条件培养基中加入NaCl使其高渗(终浓度增加150mmol/L),以快速可逆地阻断网格蛋白包被小窝的形成;将高渗条件培养基孵育HepG2细胞3小时,采用流式细胞术检测LDL内吞情况及细胞表面LDLR含量,同时检测红色荧光标记的PCSK9(WT-PCSK9-Red)的内吞效率。结果解读:高渗介质下,LDL的内吞被抑制至12%(n=3,P<0.01),但D374Y-PCSK9组的细胞表面LDLR仍显著低于空质粒组(n=3,P<0.05);WT-PCSK9-Red的内吞在高渗介质下略有降低但仍可进行(n=3,P<0.05)(图5、6),提示PCSK9介导的LDLR降解不依赖网格蛋白介导的内吞途径,PCSK9可通过非经典内吞方式进入细胞。产品关联:文献未提及具体实验产品,领域常规使用荧光标记试剂、流式细胞仪等。
3.5 诺考达唑与氯化铵实验验证胞内降解途径
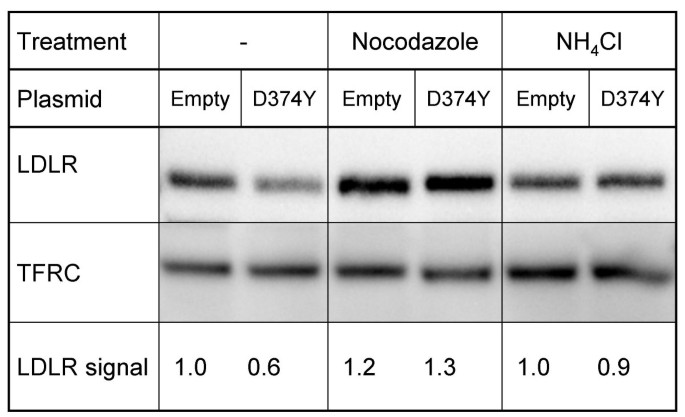
实验目的:解析PCSK9介导的LDLR降解所需的胞内运输及区室条件。方法细节:在HepG2细胞培养基中加入诺考达唑(20μg/ml,破坏微管)或氯化铵(10mM,升高酸性区室pH)预处理30分钟,随后更换含PCSK9及相应抑制剂的条件培养基继续孵育3小时,通过免疫印迹检测LDLR的含量变化。结果解读:免疫印迹结果显示,诺考达唑和氯化铵均可显著抑制PCSK9介导的LDLR降解(图7),说明LDLR的降解需要依赖微管的胞内运输,且发生在酸性胞内区室(如溶酶体)。产品关联:文献未提及具体实验产品,领域常规使用细胞抑制剂、免疫印迹抗体等。
4. Biomarker研究及发现成果解析
本文涉及的Biomarker为PCSK9,作为脂蛋白代谢调控的关键功能分子,其通过特定机制降解LDLR,影响血胆固醇水平,具有重要的疾病关联与药物靶点价值。
Biomarker定位:PCSK9属于功能型Biomarker,其筛选与验证逻辑基于前期领域内对PCSK9与LDLR关联的发现,本文通过细胞实验系统验证其作用机制。研究过程详述:PCSK9来源于转染HepG2细胞的分泌产物,验证方法包括亲和层析、凝胶过滤、免疫印迹、流式细胞术等;特异性方面,PCSK9仅在肝、肾细胞中发挥降解LDLR的效应,本文以HepG2肝细胞为模型,敏感性上,D374Y功能获得性突变体的降解活性显著高于野生型,可抑制73%±3%的LDL内吞(n=4,P<0.0001)。核心成果提炼:PCSK9直接介导LDLR的胞内降解,无需胞外其他分泌蛋白参与;降解过程不依赖网格蛋白介导的内吞,需要微管运输至酸性胞内区室;创新性在于首次完整解析了PCSK9降解LDLR的关键机制,为靶向PCSK9的降脂药物开发提供了核心理论基础,相关统计学结果包括多个实验的P值(如P<0.01、P<0.001、P<0.0001),样本量为3或4。