1. 领域背景与文献引入
文献英文标题:NCBoost classifies pathogenic non-coding variants in Mendelian diseases through supervised learning on purifying selection signals in humans;发表期刊:Genome Biology;影响因子:17.906(2023年);研究领域:孟德尔疾病非编码遗传变异致病性预测。
领域共识:孟德尔疾病是由单基因突变引起的遗传性疾病,目前已临床识别超过4000种,仅美国就有超过2500万患者。全外显子测序(WES)在孟德尔疾病诊断中的应用取得了一定进展,但不同疾病类型的诊断率差异极大,从低于15%(如先天性膈疝)到超过70%(如纤毛运动障碍),约50%的孟德尔疾病仍未找到致病基因或变异。随着全基因组测序(WGS)的普及,非编码区变异被认为是解释这些未解决病例的关键,因为全基因组关联研究(GWAS)发现大多数疾病相关变异位于非编码区,提示调控元件在遗传疾病中的重要作用。然而,非编码区存在大量罕见和 singleton 变异,需要计算方法来优先排序候选变异,以便后续临床和实验验证。
当前研究热点方向包括开发基于机器学习的非编码变异致病性预测工具,整合跨物种保守性、表观遗传特征、人类种群遗传特征等多种信息。未解决的核心问题是现有方法在孟德尔疾病非编码变异预测中的准确性有限,存在严重的位置偏倚,例如5"UTR区域的非致病性常见变异得分与3"UTR区域的致病性变异得分无显著差异,甚至高于内含子区域的致病性变异得分,导致变异优先级排序错误。针对这一问题,本研究旨在开发一种更准确的非编码变异致病性预测工具NCBoost,通过整合人类近期和正在进行的净化选择信号,克服现有方法的位置偏倚,提高孟德尔疾病非编码变异的预测性能,为孟德尔疾病的分子诊断提供新的技术支持。
2. 文献综述解析
作者对领域内现有研究的分类维度主要为特征类型(跨物种保守性、表观遗传特征、人类种群遗传特征)和针对的疾病类型(常见疾病、孟德尔疾病)。
现有研究中,支持观点认为跨物种保守性和表观遗传特征能够在一定程度上区分功能相关的非编码变异,例如基于跨物种保守性的方法(如PhastCons、PhyloP)已被广泛用于识别具有功能的基因组区域,表观遗传特征(如组蛋白修饰、开放染色质)能够反映调控元件的活性。技术方法优势方面,ReMM是首个专门针对孟德尔疾病非编码变异的预测工具,整合了8种跨物种保守性特征、4种GC/CpG特征和8种表观遗传特征,通过随机森林模型训练,在孟德尔疾病变异优先排序中显示出一定价值;基于深度学习的方法(如DeepSEA)能够从DNA序列中学习调控元件的特征,预测非编码变异的功能影响。局限性方面,现有方法在孟德尔疾病非编码变异预测中的准确性中等,主要依赖跨物种保守性特征,表观遗传特征的贡献较小;存在严重的位置偏倚,不同基因组区域的变异得分分布存在系统偏差,导致非致病性变异得分被高估,致病性变异得分被低估;大多数方法是针对常见疾病相关变异开发的,在孟德尔疾病大效应非编码变异预测中的适用性有限。
通过对比现有研究的未解决问题,本研究的创新价值凸显:首次全面整合人类近期和正在进行的净化选择信号,结合跨物种保守性、基因水平特征和序列上下文,使用梯度提升树的监督学习方法构建NCBoost模型,有效克服了现有方法的位置偏倚;在多种测试场景下(交叉验证、独立验证集、模拟个体基因组)均显著优于现有方法,包括专门针对孟德尔疾病开发的ReMM方法;首次证明人类近期和正在进行的净化选择信号在孟德尔疾病非编码变异致病性预测中的重要作用,为非编码变异致病性预测提供了新的特征维度。
3. 研究思路总结与详细解析
本研究的整体框架为:以构建准确的孟德尔疾病非编码单核苷酸变异(SNV)致病性预测工具为目标,核心科学问题是如何整合人类近期和正在进行的净化选择信号,克服现有方法的位置偏倚,提高非编码变异致病性预测的准确性;技术路线遵循“数据构建→特征提取→模型训练→性能评估→独立验证→应用拓展”的闭环逻辑,通过多步验证确保模型的可靠性和实用性。
3.1 高置信度非编码致病性变异集的构建与验证
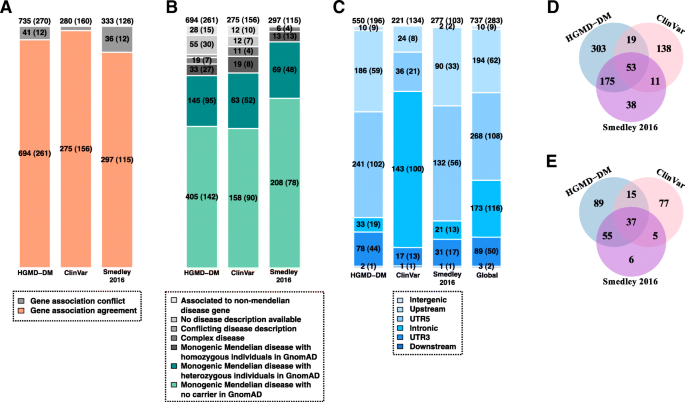
实验目的是构建用于模型训练和测试的高置信度孟德尔疾病非编码致病性变异集,确保数据的可靠性和代表性。方法细节包括从HGMD-DM、ClinVar和Smedley’2016三个数据库获取非编码致病性变异,经过多步严格过滤:排除外显子和剪接位点附近(10bp内)的变异,排除与非编码RNA相关的变异,手动校正基因注释不一致的情况,排除在GnomAD数据库中存在纯合子的变异,最终保留737个与283个孟德尔疾病基因相关的非编码SNV,这些变异分布在5"UTR(36%)、1kb上游转录起始位点(TSS,26%)、内含子(23%)、3"UTR(12%)等区域。结果解读显示,该变异集显著富集于单倍剂量不足基因(比值比OR=2.93,单侧Fisher检验P=1.279e−09)和不耐受杂合截断的基因(OR=1.21,P=0.041),符合孟德尔疾病基因的功能特征,说明构建的变异集具有较高的可靠性。文献未提及具体实验产品,领域常规使用ANNOVAR等注释工具、R等统计分析软件完成数据处理。
3.2 特征提取与单特征性能评估
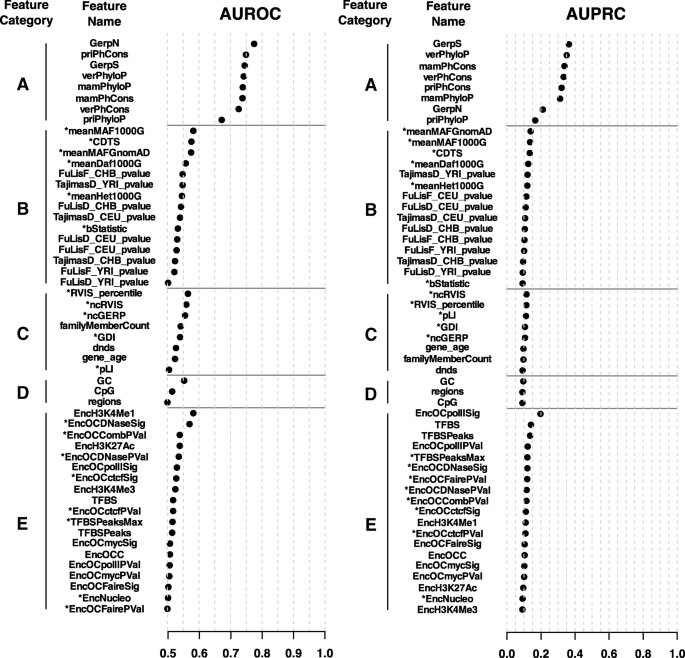
实验目的是提取用于模型训练的多种特征,并评估单个特征区分致病性与非致病性变异的能力,为后续特征组合提供依据。方法细节包括提取五类特征:A类为跨物种序列保守性特征(如PhastCons、PhyloP、GerpN/S),B类为人类近期和正在进行的净化选择信号(如Tajima’s D、Fu and Li’s F/D、CDTS、平均杂合度),C类为基因水平特征(如dn/ds、pLI、GDI),D类为序列上下文特征(GC/CpG含量、变异所在区域),E类为表观遗传特征(如组蛋白修饰、开放染色质、转录因子结合位点)。使用受试者工作特征曲线下面积(AUROC)和精确召回曲线下面积(AUPRC)评估单个特征在区分737个致病性变异和7370个匹配区域的常见非致病性变异的性能。结果解读显示,单个特征中跨物种保守性特征的预测性能最优(AUROC>0.7),而其他特征单独使用时性能较差(AUROC<0.6,AUPRC<0.2),说明单个特征难以有效区分致病性与非致病性变异,需要整合多种特征才能提高预测性能。文献未提及具体实验产品,领域常规使用CADD、GWAVA等工具提取预计算特征,使用R进行统计分析和绘图。
3.3 NCBoost模型构建与现有方法性能对比
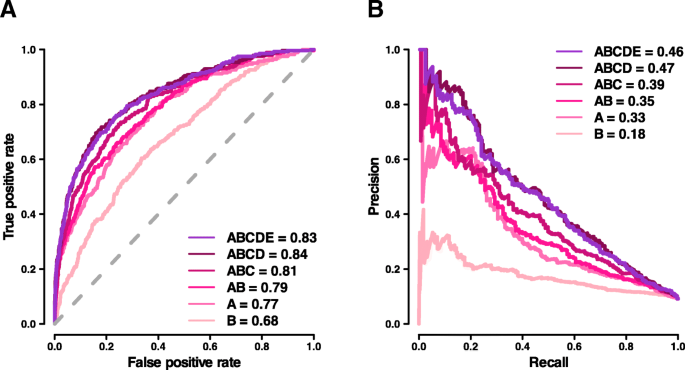
实验目的是构建基于梯度提升树的监督学习模型NCBoost,评估不同特征组合的性能,并与现有主流方法对比,验证模型的优越性。方法细节包括使用XGBoost训练模型,采用10个独立模型的bundle策略,每个模型排除1/10的基因组分区,避免基因水平的过拟合;训练集为283个致病性变异(每个基因随机选择1个)和2830个匹配区域的常见非致病性变异(每个基因最多1个)。评估六种特征组合(A、B、A+B、A+B+C、A+B+C+D、A+B+C+D+E)的性能,然后在相同数据集上与六种现有方法(CADD、DeepSEA、Eigen、Eigen-PC、FunSeq2、ReMM)对比性能。结果解读显示,包含A+B+C+D特征的模型性能最优,AUROC=0.84,AUPRC=0.47,比仅使用A类特征的模型分别提高9%和42%;特征重要性分析显示,跨物种保守性特征(累计重要性42%)和人类净化选择信号(累计重要性33%)对模型性能的贡献最大。NCBoost在AUROC和AUPRC上均显著优于所有现有方法,相对ReMM分别提高10%和34%,说明整合人类净化选择信号能够显著提高模型性能。文献未提及具体实验产品,领域常规使用XGBoost库(R或Python版本)进行模型训练,使用R进行统计分析和绘图。
3.4 独立验证与模拟个体基因组变异优先排序
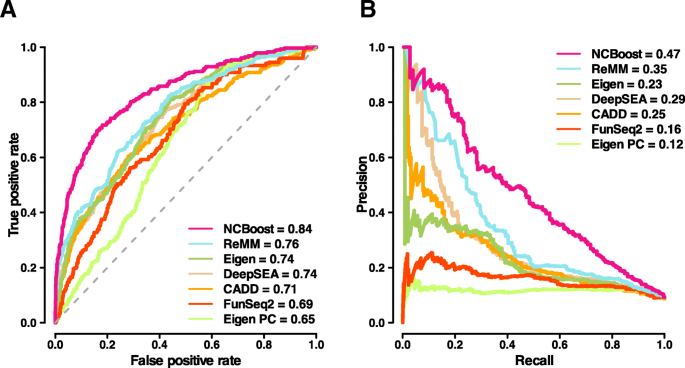
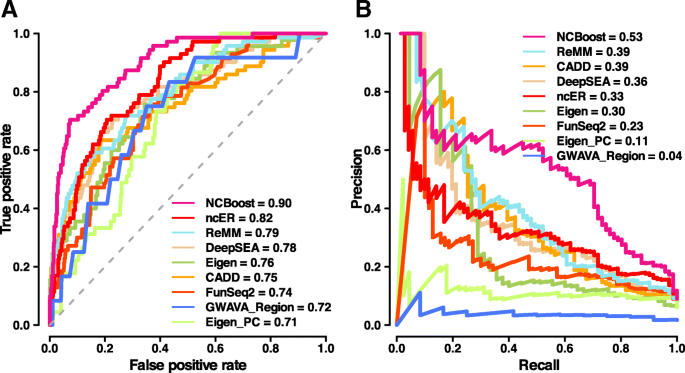
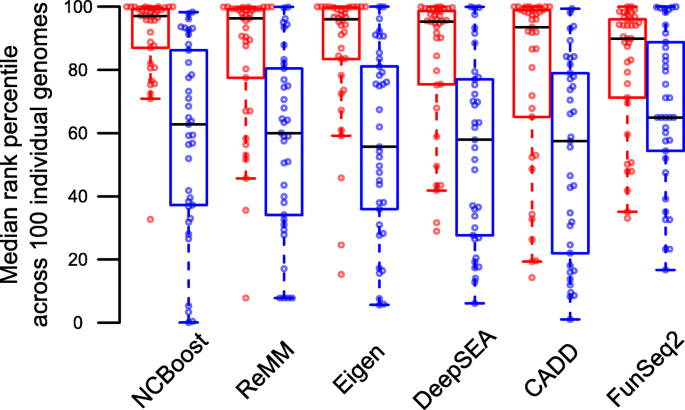
实验目的是在独立验证集和模拟个体基因组中验证NCBoost的性能,评估其在实际临床场景中的应用价值。方法细节包括:独立验证集包含70个新报道的非编码致病性变异和700个匹配区域的常见非致病性变异;模拟个体基因组是将37个致病性变异和对应的37个非致病性变异分别插入100个1000 Genomes Project的健康个体基因组中,然后对每个个体的非编码变异进行评分并排序。结果解读显示,在独立验证集中,NCBoost的AUROC=0.90,AUPRC=0.53,显著优于所有现有方法,性能与交叉验证结果一致,说明模型不存在过拟合;在模拟个体基因组中,NCBoost的致病性变异的中位排名百分位数为97.04%,显著高于其他方法(除Eigen外,P<0.05),且致病性变异与非致病性变异的排名百分位数差异最显著(P=8.04e−7),说明NCBoost能够在个体基因组中有效优先排序致病性非编码变异,具有较高的临床应用潜力。文献未提及具体实验产品,领域常规使用PLINK等工具处理基因组变异数据,使用R进行统计分析和绘图。
3.5 孟德尔疾病基因非编码区全基因组评分
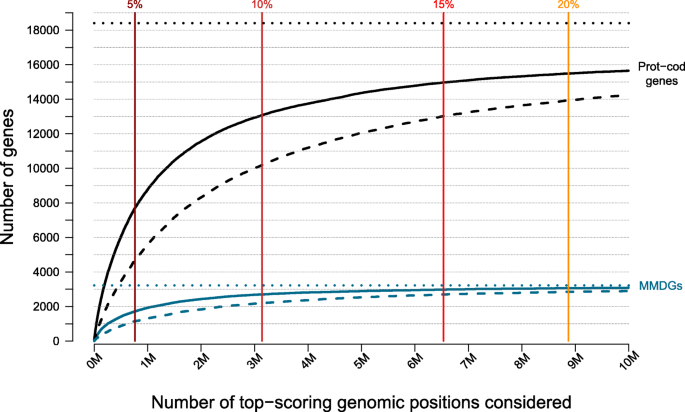
实验目的是评估NCBoost在全基因组范围内对孟德尔疾病基因非编码区变异的致病性评分能力,识别潜在的致病性变异区域。方法细节包括对3223个孟德尔疾病基因的非编码区(内含子、5"UTR、3"UTR、上下游1kb)的189,829,714个基因组位置进行NCBoost评分,统计得分高于高置信度致病性变异前5%和前10%的位置和基因数量。结果解读显示,共有261,507个位置得分高于前5%,对应1715个孟德尔疾病基因;980,219个位置得分高于前10%,对应2699个孟德尔疾病基因。这些基因显著富集于孟德尔疾病基因(OR=1.27,P=3.78e−13;OR=1.18,P=8.81e−09),说明NCBoost能够有效识别孟德尔疾病基因的潜在致病性非编码区域,为孟德尔疾病的分子诊断提供新的候选变异。文献未提及具体实验产品,领域常规使用生物信息学脚本完成全基因组评分,使用R进行统计分析和绘图。
4. Biomarker研究及发现成果
本研究的核心是开发的NCBoost模型作为一种“虚拟Biomarker”,用于预测孟德尔疾病非编码SNV的致病性,其筛选逻辑是整合跨物种保守性、人类近期和正在进行的净化选择信号、基因水平特征和序列上下文,通过监督学习训练模型,对未知变异进行评分,得分越高则致病性越高。
研究过程中,模型的特征来源包括公共数据库(如GnomAD、1000 Genomes Project、ENCODE)的基因组变异数据、进化保守性数据、表观遗传数据;验证方法包括十折交叉验证、独立验证集验证、模拟个体基因组验证;特异性与敏感性方面,在交叉验证中AUROC=0.84,AUPRC=0.47,说明模型能够有效区分致病性与非致病性变异;在独立验证集中AUROC=0.90,AUPRC=0.53,进一步验证了模型的特异性和敏感性;在模拟个体基因组中,致病性变异的中位排名百分位数为97.04%,说明模型能够在大量背景变异中准确识别致病性变异。
核心成果提炼方面,NCBoost模型能够有效克服现有方法的位置偏倚,在多种测试场景下均显著优于现有非编码变异致病性预测工具;首次证明整合人类近期和正在进行的净化选择信号能够显著提高孟德尔疾病非编码变异致病性预测的准确性,为非编码变异致病性预测提供了新的特征维度;模型可用于全基因组范围内孟德尔疾病基因非编码区的潜在致病性变异识别,为孟德尔疾病的分子诊断提供新的工具。统计学结果显示,所有性能指标均具有显著的统计学差异,例如模拟个体基因组中致病性与非致病性变异排名百分位数的差异(P=8.04e−7),NCBoost与现有方法的性能差异(P<0.05,文献未明确提供具体P值,但性能提升幅度具有统计学意义)。