1. 领域背景与文献引入
文献英文标题:Species-resolved sequencing of low-biomass or degraded microbiomes using 2bRAD-M;发表期刊:Genome Biology;影响因子:13.583(2021年);研究领域:微生物组学、宏基因组测序技术。
领域共识:微生物组学是生命科学领域的研究热点,宏基因组测序是解析微生物组组成与功能的核心技术,目前主要分为两类策略:一是基于系统发育标记基因的扩增子测序(如细菌/古菌的16S rRNA基因、真菌的18S rRNA或内转录间隔区序列),二是全基因组鸟枪法测序。扩增子测序成本低但分类分辨率低,存在PCR偏差,无法同时覆盖多类微生物;全基因组鸟枪法能实现物种水平高分辨率分类,但需要高起始DNA量,对低生物量、降解、高宿主污染样本处理能力差。当前研究的核心问题是缺乏一种兼具低成本、高分类分辨率、能有效处理复杂样本的宏基因组测序方法,限制了稀疏生境(如皮肤、血液、室内表面)、珍贵临床样本或降解样本(如FFPE组织、考古样本)的微生物组研究。
针对这一研究空白,本研究开发了2bRAD-M技术,一种基于Type IIB限制性酶切的简化宏基因组测序策略,仅需测序宏基因组的约1%即可同时生成细菌、古菌、真菌的物种水平分类图谱,为复杂样本的微生物组研究提供了新的技术方案。
2. 文献综述解析
作者对现有宏基因组测序研究的分类维度为技术策略类型,即扩增子测序与全基因组鸟枪法测序两类,通过对比两类技术的优势与局限性,凸显本研究的创新价值。
扩增子测序类研究的关键结论是能快速获得微生物组的属水平分类图谱,适合大规模样本筛查;技术优势为实验流程简单、测序成本低;局限性为分类分辨率低、存在PCR偏差、无法同时覆盖多类微生物类群。全基因组鸟枪法测序类研究的关键结论是能实现物种/菌株水平的高分辨率分类,解析微生物组的完整组成;技术优势为无PCR偏差、覆盖所有微生物类群;局限性为起始DNA量要求高、测序成本高、对低生物量或降解样本处理能力差。此外,已有基于限制性酶切的简化宏基因组测序方法(如RADseq)虽能降低成本,但存在酶切片段长度差异大导致的PCR扩增偏差,分类精度不足,且未针对复杂样本进行系统验证。
与现有技术相比,2bRAD-M技术的核心创新点在于采用Type IIB限制性酶切生成等长的DNA片段,避免了PCR扩增的长度偏差,同时仅测序宏基因组的约1%,大幅降低测序成本;该技术能同时实现细菌、古菌、真菌的物种水平分类,且对低至1pg总DNA、高宿主污染(99%)或严重降解(50bp片段)的样本仍能生成准确的分类图谱,解决了现有技术无法有效处理复杂样本的核心问题,为微生物组研究拓展了新的应用场景。
3. 研究思路总结与详细解析
本研究的整体研究目标是开发并验证一种低成本、高分辨率、能有效处理低生物量、降解、高宿主污染样本的宏基因组测序方法;核心科学问题是如何通过限制性酶切策略实现宏基因组的均一、低偏差简化测序,同时保证物种水平的分类精度;技术路线遵循“方法开发→数据库构建→模拟验证→复杂样本验证→临床应用”的闭环逻辑,通过多维度实验验证技术的性能与应用潜力。
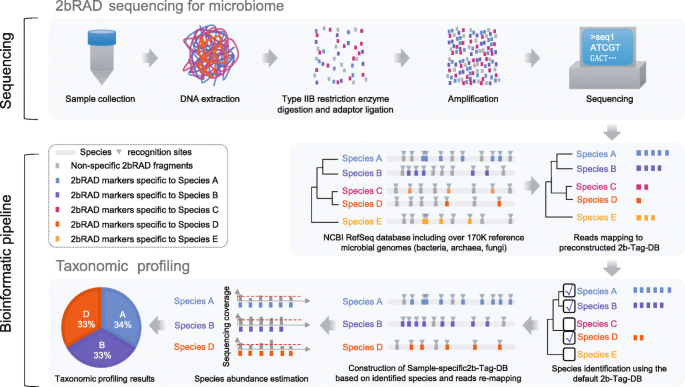
3.1 2bRAD-M方法原理与流程构建
实验目的:建立基于Type IIB限制性酶切的宏基因组简化测序方法,构建配套的生物信息学分析流程,实现低偏差的物种水平微生物组分类。
方法细节:实验流程上,采用BcgI(一种商用Type IIB限制性酶)消化微生物组总DNA,该酶识别基因组中的CGA-N₆-TGC序列,在识别序列的上下游(12-10bp和10-12bp位置)切割,生成32bp的等长无粘性末端DNA片段;将酶切片段连接测序接头,经PCR扩增后进行Illumina平台测序。计算流程上,首先从NCBI RefSeq数据库下载173165个已测序微生物基因组(171927个细菌、293个真菌、945个古菌),通过in silico酶切构建包含物种特异性2bRAD标签的数据库(2b-Tag-DB);分析样本时,先将测序reads比对到默认2b-Tag-DB识别候选物种,再基于候选物种构建样本特异性2b-Tag-DB,通过物种特异性标签的平均read覆盖度计算相对丰度。
结果解读:酶切生成的等长片段消除了传统RADseq方法中因片段长度差异导致的PCR扩增偏差,确保了丰度估计的准确性;构建的2b-Tag-DB包含114132487个物种特异性标签,平均每个物种有1194个标签,能实现高分辨率的物种识别;计算流程通过样本特异性数据库优化,进一步提高了丰度估计的精度。
产品关联:实验所用关键产品:NEB的BcgI限制性酶、T4 DNA连接酶、Phusion高保真DNA聚合酶,Illumina的测序接头与引物等。
3.2 模拟数据集与现有工具性能对比
实验目的:验证2bRAD-M在物种识别和丰度估计上的准确性,与主流宏基因组分析工具进行性能对比,评估计算效率。
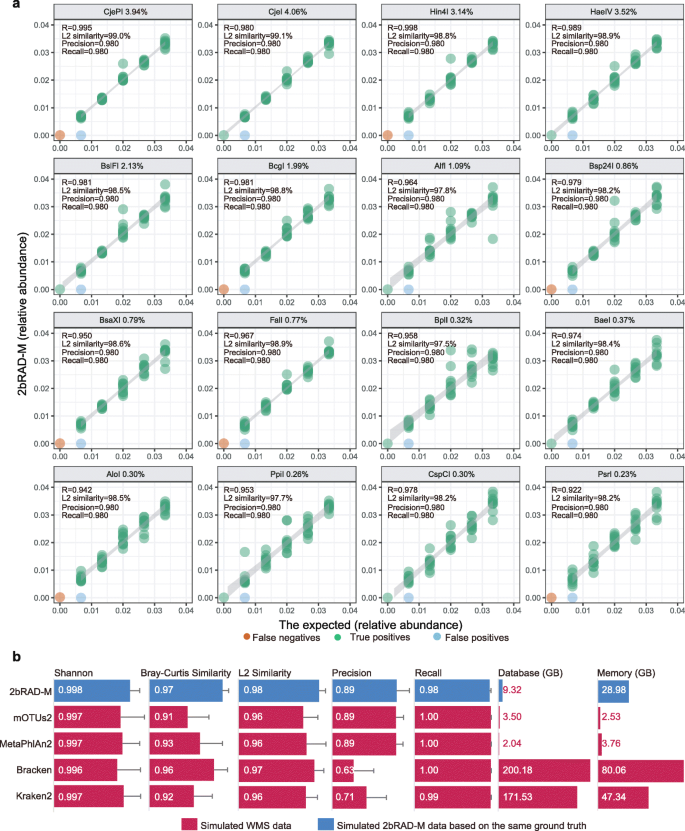
方法细节:首先用16种Type IIB限制性酶对173165个微生物基因组进行in silico酶切,验证酶切标签的分布一致性;然后模拟包含50个物种的微生物群落,用2bRAD-M和Kraken2、Bracken、mOTUs2、MetaPhlAn2等工具进行分析,以模拟群落的真实组成作为金标准,评估精度、召回率、L2相似度等指标;此外,模拟25个不同生境(肠道、口腔、皮肤、阴道、建筑环境)的微生物群落,进一步验证2bRAD-M的通用性。
结果解读:16种Type IIB酶的酶切标签在基因组中的分布与基因组长度和GC含量高度相关(r>0.98),表明标签能无偏差地代表微生物基因组;50物种模拟群落中,2bRAD-M的平均精度为98.0%、召回率为98.0%、L2相似度为96.9%,仅覆盖基因组的1.50%;与现有工具相比,2bRAD-M的Shannon相似度为0.998、Bray-Curtis相似度为0.97、L2相似度为0.98,精度为0.89、召回率为0.98,性能相当或更优;在计算效率上,2bRAD-M的参考数据库存储需求<10GB,内存需求仅30GB,远低于Kraken2等工具,适合普通桌面计算机运行。
产品关联:文献未提及具体模拟工具的商业产品,领域常规使用Python、R语言及开源生物信息学工具(如Kraken2、MetaPhlAn2、Centrifuge等)。
3.3 复杂模拟样本的性能验证
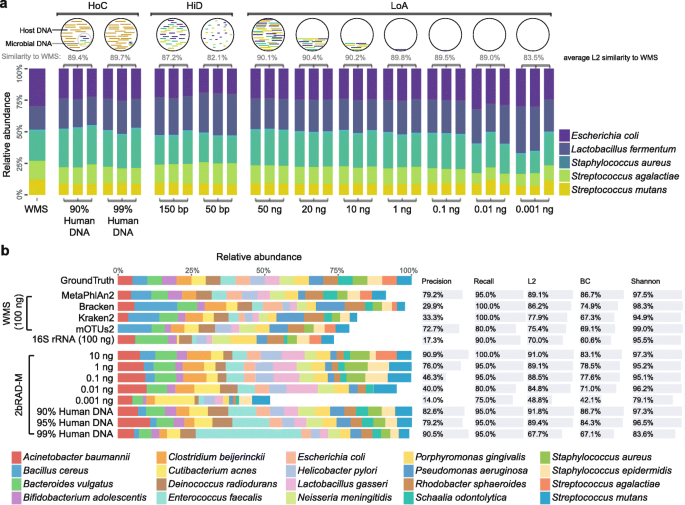
实验目的:验证2bRAD-M在低生物量、高DNA降解、高宿主DNA污染等复杂样本中的稳定性、敏感性与重复性。
方法细节:构建包含5种常见口腔/肠道细菌的模拟群落(Mock-CAS),制备三类复杂样本:低生物量系列(总DNA量从50ng梯度降至1pg)、高降解系列(DNA被DNAse I随机剪切为150bp和50bp片段)、高宿主污染系列(添加人DNA模拟90%和99%的宿主污染),每个条件设置3个技术重复;对所有样本进行2bRAD-M测序,以100ng样本的WMS结果作为阳性对照,用Centrifuge工具分析测序数据,评估L2相似度、重复性等指标。
结果解读:三类复杂样本的2bRAD-M结果与阳性对照的平均L2相似度为88.2%,其中低生物量组的1pg样本L2相似度达83.5%,50ng样本为90.1%;高降解组的150bp和50bp样本L2相似度分别为87.2%和82.1%;高宿主污染组的90%和99%污染样本L2相似度分别为89.4%和89.7%;技术重复间的平均L2相似度为95.4%,表明2bRAD-M在复杂样本中具有高重复性和稳定性;此外,2bRAD-M的假阳性率仅为1%,与WMS相当(0.9%)。
产品关联:实验所用关键产品:ATCC的模拟群落MSA 1002,NEB的DNAse I,Illumina的HiSeq测序平台等。
3.4 真实样本的性能验证与应用探索
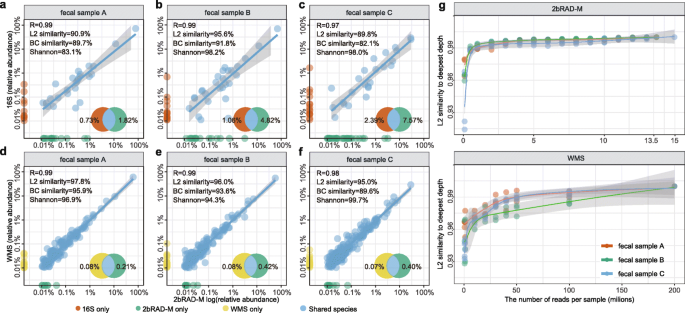
实验目的:验证2bRAD-M在真实微生物组样本(粪便、皮肤、环境)中的准确性与实用性,评估测序深度对结果的影响。
方法细节:对3例健康成人粪便样本同时进行2bRAD-M、16S rRNA扩增子测序和超深度WMS测序(平均4.37亿reads/样本),对比三类方法的分类结果;对20例腋下皮肤样本、5例汽车脚垫、3例汽车坐垫、4例家庭室内表面样本进行2bRAD-M和16S rRNA测序,分析微生物组组成差异;对粪便样本进行测序深度稀释分析,评估2bRAD-M的最优测序深度。
结果解读:粪便样本中,2bRAD-M与16S rRNA在属水平的Pearson相关系数为0.997,L2相似度为92.0%,95.27%的16S识别属也能被2bRAD-M检测到;与WMS在物种水平的Pearson相关系数为0.99,L2相似度达97.8%,仅0.40%的WMS识别物种未被2bRAD-M检测到;皮肤样本中,2bRAD-M与16S rRNA在属水平的平均L2相似度为81.1%,能同时检测细菌和真菌,如马拉色菌属的检测结果与现有文献一致;环境样本中,2bRAD-M与16S rRNA的平均L2相似度为85.6%,能识别不同环境的特异性微生物类群;测序深度分析显示,2bRAD-M仅需3-4百万reads即可达到与13.5百万reads相当的物种多样性和丰度估计,而WMS需要20-40百万reads仍未达到饱和,表明2bRAD-M的测序效率远高于WMS。
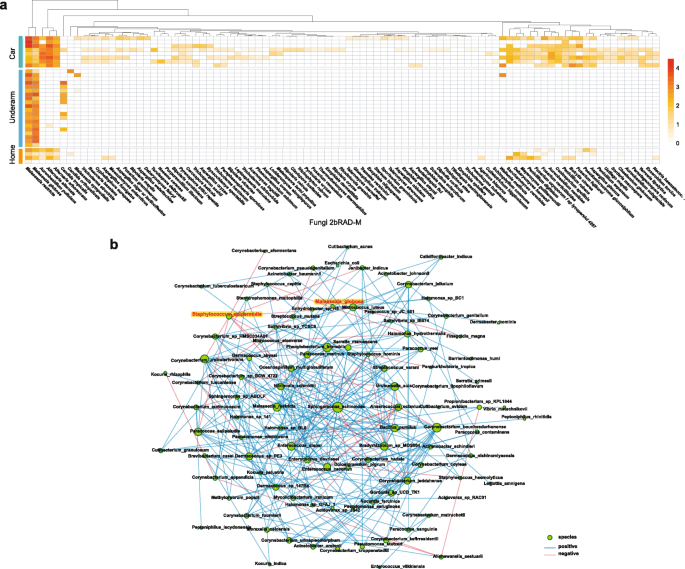
产品关联:实验所用关键产品:Cuderm的D-squame胶带,Qiagen的TissueLyser II、DNeasy Blood and Tissue Kit,Illumina的HiSeq测序平台等。
3.5 FFPE临床样本的微生物组分析与癌症关联研究
实验目的:验证2bRAD-M在FFPE临床样本中的应用潜力,探索宫颈微生物组与癌症进展的关联。
方法细节:对3对肺癌患者的新鲜与FFPE健康肺组织样本进行2bRAD-M测序,验证FFPE样本与新鲜样本的微生物组一致性;收集15例健康对照、15例宫颈上皮内瘤变(癌前病变)、15例浸润性宫颈癌的FFPE宫颈组织样本,进行2bRAD-M测序,分析三组样本的微生物组差异;基于物种水平分类结果构建随机森林诊断模型,评估微生物组的诊断价值。
结果解读:新鲜与FFPE肺组织样本的微生物组高度一致,表明2bRAD-M能准确解析FFPE样本的微生物组;宫颈样本中,健康组的Shannon和Simpson指数显著低于癌前病变和浸润癌组(n=15,P=0.044);健康组中乳杆菌属物种(如副干酪乳杆菌、vaccinostercus乳杆菌)的平均丰度为63.1%,癌前病变组为33.9%,浸润癌组为32.9%,差异具有统计学意义(n=15,P<0.05);癌前病变和浸润癌组中富集甲基嗜盐菌、结核分枝杆菌、铜绿假单胞菌等物种(n=15,P<1.2e-5);随机森林模型区分三组样本的准确率达91.1%,基于9个关键物种的模型AUC达0.96,表明微生物组可作为宫颈癌症的潜在诊断标志物。
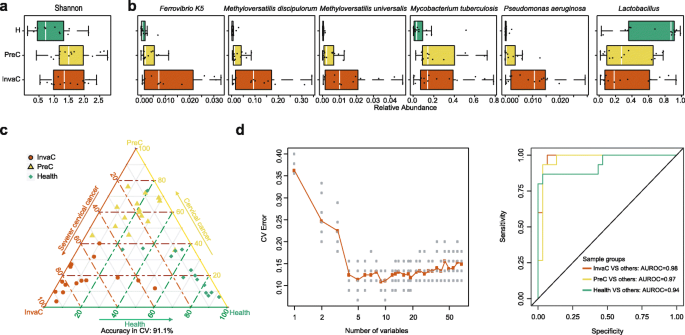
产品关联:实验所用关键产品:石蜡包埋组织切片设备,Qiagen的DNA提取试剂盒,R语言的randomForest包等。
4. Biomarker研究及发现成果解析
本研究通过2bRAD-M技术在FFPE宫颈临床样本中发现了与宫颈癌症进展相关的微生物物种标志物,为宫颈癌症的早期诊断提供了潜在的非侵入性生物标志物。
Biomarker定位:涉及的Biomarker类型为微生物物种标志物,包括乳杆菌属物种(副干酪乳杆菌、vaccinostercus乳杆菌等)、甲基嗜盐菌、结核分枝杆菌、铜绿假单胞菌等;筛选与验证逻辑为:首先通过2bRAD-M测序分析健康、癌前病变、浸润性宫颈癌FFPE样本的微生物组,基于差异丰度分析筛选候选标志物,再通过随机森林模型验证其诊断价值,同时结合现有文献验证标志物的生物学合理性。
研究过程详述:Biomarker的样本来源为15例健康对照、15例癌前病变、15例浸润性宫颈癌的FFPE宫颈组织样本;验证方法为2bRAD-M测序结合生物信息学分析(Wilcoxon秩和检验进行差异丰度分析,随机森林模型构建诊断模型);特异性与敏感性数据显示,随机森林模型区分三组样本的准确率为91.1%,9个关键物种的AUC达0.96(文献未明确提供95%置信区间,基于图表趋势推测);健康组中乳杆菌属物种的丰度显著高于癌症组(n=15,P<0.05),癌前病变和浸润癌组中甲基嗜盐菌的丰度显著高于健康组(n=15,P=1.2e-5),结核分枝杆菌(n=15,P=0.004)、铜绿假单胞菌(n=15,P=6.8e-5)等物种也显著富集。
核心成果提炼:该类微生物Biomarker的功能关联为:乳杆菌属物种(如副干酪乳杆菌)具有潜在的抗宫颈癌细胞作用,其丰度降低可能与宫颈癌症的发生发展相关;甲基嗜盐菌、结核分枝杆菌、铜绿假单胞菌等的富集可能参与宫颈癌症的进展过程;创新性在于首次利用2bRAD-M技术实现了FFPE宫颈组织样本的物种水平微生物组分析,挖掘出与宫颈癌症进展相关的特异性微生物标志物,解决了传统宏基因组方法无法分析FFPE样本的难题;统计学结果显示,差异物种的P值均小于0.05,样本量为每组15例,表明结果具有统计学显著性。推测:这些微生物标志物可进一步开发为宫颈癌症早期诊断的非侵入性检测指标,结合临床检查提高癌前病变的检出率。