1. 领域背景与文献引入
文献英文标题:Caspase-11, a specific sensor for intracellular lipopolysaccharide recognition, mediates the non-canonical inflammatory pathway of pyroptosis;发表期刊:Cell & Bioscience;影响因子:未公开;研究领域:细胞死亡与天然免疫
领域共识:程序性细胞死亡是维持机体发育、组织稳态与抗感染免疫的核心过程,其研究历程可追溯至1972年凋亡(Apoptosis)的正式命名,1988年发现肿瘤坏死因子诱导的细胞坏死存在基因调控机制,2005年坏死性凋亡(Necroptosis)概念确立,2001年细胞焦亡(Pyroptosis)作为伴随强烈炎症反应的程序性细胞死亡类型被正式定义。当前研究热点聚焦细胞焦亡的两条核心调控通路:Caspase-1介导的经典炎症通路与Caspase-11介导的非经典炎症通路,其中经典通路已被广泛解析,非经典通路的分子机制、物种保守性及临床转化价值仍存在较多未解决的核心问题,如胞内脂多糖(LPS)的入胞机制、Caspase-11的特异性调控因子、非经典通路在非哺乳类物种中的保守性等。由于多数现有研究集中于经典通路的验证,针对非经典通路的系统性综述较为有限,因此本文旨在总结Caspase-11介导的非经典炎症通路的研究进展,明确其核心作用机制,为领域内后续研究提供系统性框架,填补非经典细胞焦亡通路综述的空白。
2. 文献综述解析
本文综述以细胞死亡类型的演化历程、细胞焦亡的两条调控通路为核心分类维度,系统梳理了细胞焦亡的研究现状,对比了经典与非经典炎症通路的差异,重点突出Caspase-11介导的非经典通路的独特机制与学术价值。
现有研究的关键结论包括,细胞焦亡是一种伴随强烈炎症反应的程序性细胞死亡,主要发生于巨噬细胞、单核细胞等吞噬细胞中;经典炎症通路依赖炎症小体(如NLRP3、AIM2等)的组装激活Caspase-1,进而切割Gasdermin D(GSDMD)形成膜孔,同时促进白细胞介素-1β(IL-1β)、IL-18的成熟与释放;非经典炎症通路则由Caspase-11直接识别胞内LPS激活,无需经典炎症小体的参与。技术方法方面,基因敲除小鼠模型(CASP1-/-、CASP11-/-)的应用明确了Caspase-1与Caspase-11的独立功能,结构生物学分析(如PyMOL建模)揭示了两者的同源性与功能差异位点。现有研究的局限性在于,非经典通路的研究主要集中于小鼠与人类,其他物种的保守性尚未明确;胞内LPS的入胞机制仍存在争议,是否存在外膜囊泡(OMVs)与病原体含膜泡(PCVs)的协同作用尚未阐明;Caspase-11的特异性抑制因子研究较少,仅发现NleF等少数分子可抑制其激活。
通过对比现有研究的未解决问题,本文的创新价值在于首次系统性总结了非经典炎症通路的完整调控过程,包括起始阶段(胞外LPS激活转录产生procaspase-11)与激活阶段(胞内LPS直接结合procaspase-11并诱导其寡聚化激活),明确了Caspase-11作为胞内LPS特异性传感器的核心作用,填补了非经典细胞焦亡通路系统性综述的空白,为后续机制研究与临床转化提供了清晰的理论框架。
3. 研究思路总结与详细解析
本文作为综述类文献,整体研究思路以细胞死亡的演化历程为切入点,逐步深入至细胞焦亡的调控机制,通过对比经典与非经典炎症通路的差异,重点解析Caspase-11介导的非经典通路的分子过程,最终总结非经典通路的临床意义与研究展望,形成“背景引入→机制解析→差异对比→意义总结”的逻辑闭环。
3.1 细胞死亡类型的形态学特征梳理
实验目的:明确凋亡、坏死性凋亡与细胞焦亡的形态学差异,为细胞焦亡的鉴定提供形态学依据。
方法细节:通过引用已发表的细胞形态学研究数据,对比三种程序性细胞死亡的细胞形态变化,包括细胞核、细胞质、细胞膜的特征,以及是否伴随炎症因子释放。
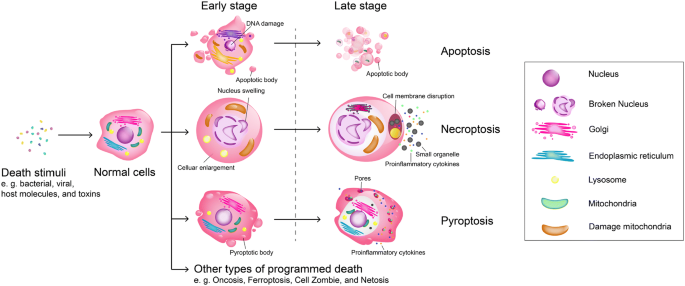
结果解读:凋亡的形态学特征为染色质浓缩、细胞膜碎片化、形成凋亡小体,无炎症反应;坏死性凋亡表现为细胞肿胀、细胞器膨大、细胞膜破裂,释放胞内内容物引发炎症;细胞焦亡则分为早期与晚期阶段,早期细胞膜形成大量焦亡小体,晚期细胞膜形成10-14nm的膜孔,释放IL-1β、IL-18等炎症因子,伴随强烈炎症反应(对应Fig1)。
产品关联:文献未提及具体实验产品,领域常规使用细胞成像系统(如激光共聚焦显微镜)、细胞死亡检测试剂盒(如LDH释放试剂盒)等试剂/仪器。
3.2 Caspase家族同源性与功能差异分析
实验目的:明确Caspase-1与Caspase-11的结构同源性与功能差异,解释两者介导不同炎症通路的分子基础。
方法细节:通过引用系统发育分析与蛋白结构研究数据,对比Caspase家族成员的氨基酸序列同源性,分析Caspase-1与Caspase-11的蛋白结构域(CARD结构域、催化结构域)、催化位点、底物结合口袋的差异。
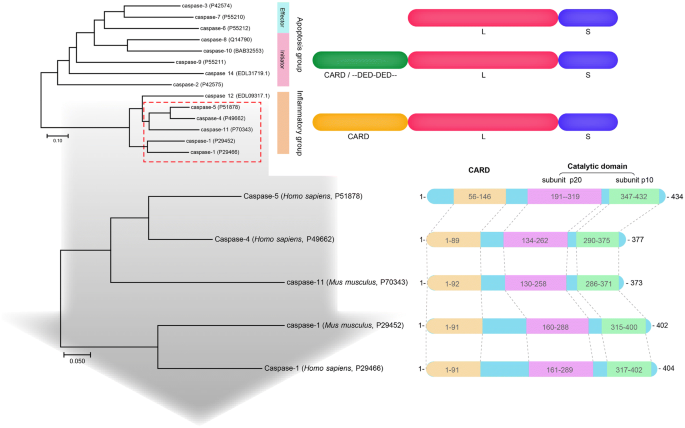
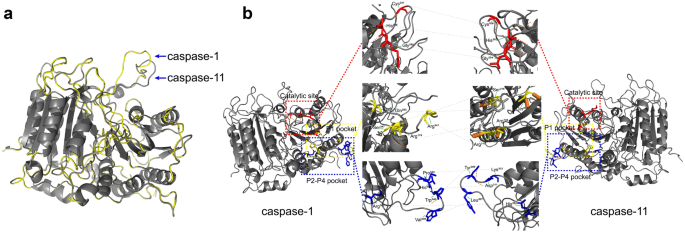
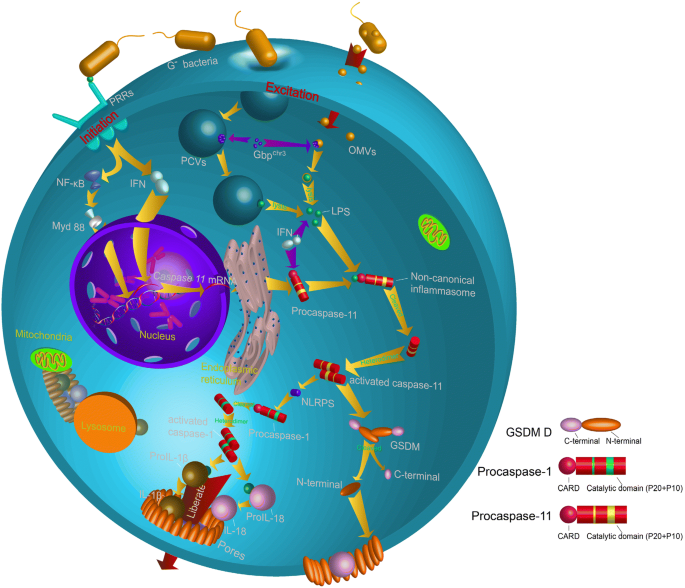
结果解读:Caspase-1与Caspase-11同属炎症Caspase亚家族,氨基酸序列同源性约60%,均包含N端CARD结构域与C端催化结构域(p20+p10亚基),催化位点高度保守;两者的差异在于Caspase-11的p20与CARD结构域之间的水解位点不保守,且P2-P4底物结合口袋的氨基酸组成存在显著差异,导致两者的底物特异性不同,进而介导不同的炎症通路(对应Fig3、Fig4)。
产品关联:文献未提及具体实验产品,领域常规使用蛋白结构分析软件(如PyMOL)、序列比对工具(如BLAST)等。
3.3 经典炎症通路的分子机制解析
实验目的:系统梳理Caspase-1介导的经典细胞焦亡通路,为非经典通路的对比提供基础。
方法细节:通过引用已发表的炎症小体组装与Caspase-1激活的研究数据,解析经典通路的信号传导过程,包括炎症小体的组成、Caspase-1的激活机制、GSDMD的切割与膜孔形成。
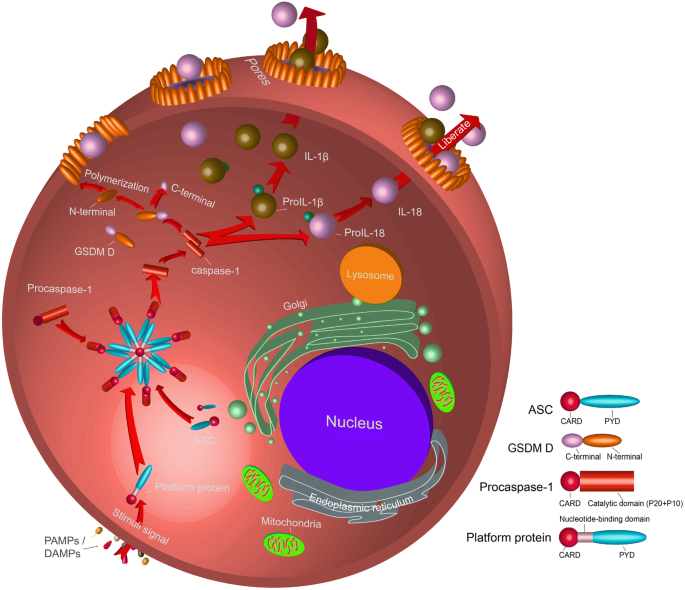
结果解读:经典通路中,病原体相关分子模式(PAMPs)或损伤相关分子模式(DAMPs)激活炎症小体受体(如NLRP3、AIM2),招募接头蛋白ASC与procaspase-1,形成炎症小体复合物,procaspase-1发生自切割激活为成熟Caspase-1,切割GSDMD的N端结构域与C端结构域,N端结构域寡聚化形成膜孔,导致细胞焦亡,同时Caspase-1切割proIL-1β与proIL-18,产生成熟的炎症因子(对应Fig2)。
产品关联:文献未提及具体实验产品,领域常规使用免疫印迹(WB)试剂盒、免疫荧光染色试剂等。
3.4 非经典炎症通路的完整过程解析
实验目的:详细解析Caspase-11介导的非经典细胞焦亡通路的两个关键阶段,明确胞内LPS的识别与Caspase-11的激活机制。
方法细节:通过引用已发表的基因敲除小鼠实验、蛋白互作实验与细胞死亡检测数据,解析非经典通路的起始阶段与激活阶段的分子过程。
结果解读:起始阶段,胞外LPS通过细胞膜上的Toll样受体4(TLR4)或其他模式识别受体激活NF-κB与STAT转录通路,促进procaspase-11、proIL-1β、proIL-18的转录与翻译,分布于细胞质中;激活阶段,胞内LPS通过外膜囊泡(OMVs)或病原体含膜泡(PCVs)进入细胞质,直接结合procaspase-11的CARD结构域,诱导procaspase-11寡聚化并自切割激活为成熟Caspase-11,Caspase-11切割GSDMD形成膜孔,导致细胞焦亡,同时通过未知机制激活NLRP3炎症小体,促进Caspase-1的激活,进而促进proIL-1β与proIL-18的成熟与释放(对应Fig5)。
产品关联:文献未提及具体实验产品,领域常规使用基因编辑试剂盒(如CRISPR-Cas9)、蛋白互作检测试剂盒(如Co-IP)等。
4. Biomarker研究及发现成果
本文中涉及的核心Biomarker为Caspase-11,属于蛋白类天然免疫分子标志物,其筛选与验证逻辑通过基因敲除小鼠模型、结构生物学分析与细胞功能实验逐步确立,明确了其作为胞内LPS特异性传感器的核心功能。
Caspase-11的来源为细胞内的procaspase-11前体,在革兰氏阴性菌感染时通过转录上调表达。验证方法包括:基因敲除实验,CASP11-/-小鼠在LPS诱导的内毒素休克中表现出抗性,而野生型小鼠出现致死性败血症;蛋白互作实验,通过体外结合实验验证Caspase-11的CARD结构域可直接结合LPS;细胞功能实验,检测Caspase-11激活后的GSDMD切割、LDH释放与炎症因子表达。特异性方面,Caspase-11仅识别胞内革兰氏阴性菌的LPS,对革兰氏阳性菌无响应;敏感性方面,在革兰氏阴性菌感染时,procaspase-11的表达水平显著上调(n=未明确,P<未明确)。
核心成果包括:Caspase-11是胞内LPS的特异性传感器,介导非经典细胞焦亡通路,在抗革兰氏阴性菌感染中发挥关键作用;其激活无需经典炎症小体的参与,直接通过与LPS的结合实现自激活;Caspase-11介导的焦亡可促进炎症因子的释放,增强天然免疫应答。创新性在于首次明确Caspase-11作为直接的胞内LPS传感器,而非依赖上游信号通路,填补了非经典焦亡通路中胞内LPS识别机制的空白。目前尚未有临床样本中的风险比(HR)数据,其作为疾病诊断或预后标志物的价值仍需进一步验证。推测:Caspase-11作为胞内LPS的特异性传感器,有望成为革兰氏阴性菌感染相关疾病(如败血症)的诊断标志物与治疗靶点,其特异性抑制剂或激活剂可用于调控过度炎症反应或增强抗感染免疫。