1. 领域背景与文献
文献英文标题:Characterisation of AGBL5 knockout in ARPE19 cells reveals rescue of ciliary defects by AGBL5 overexpression or TTLL5 knockdown;发表期刊:BMC Medical Genetics;影响因子:3.5(2023年);研究领域:视网膜纤毛病/视网膜色素变性
视网膜色素变性(RP)是最常见的遗传性视网膜退行性疾病之一,AGBL5(又称CCP5)作为微管蛋白去谷氨酰化酶,其突变与非综合征型RP的关联于2015年首次被报道,目前已被纳入英国NHS视网膜疾病基因筛查面板,但针对AGBL5相关RP仍缺乏有效治疗手段。领域共识:微管蛋白谷氨酰化修饰是调控纤毛形成与稳定性的关键机制,AGBL5作为主要的去谷氨酰化酶,其功能异常会导致纤毛缺陷,进而引发视网膜变性,但AGBL5在人类视网膜细胞中的具体作用机制尚未明确,现有研究多基于斑马鱼、小鼠等动物模型,缺乏人类细胞模型的深入机制解析与治疗策略探索。本文旨在利用人类视网膜色素上皮(ARPE19)细胞模型,构建AGBL5功能缺失的细胞系,系统表征其致病表型,并探索潜在的表型拯救策略,为AGBL5相关RP的机制研究与治疗开发提供实验依据。
2. 文献综述解析
作者从AGBL5的分子特征、酶学功能、动物模型研究及人类疾病关联四个维度对现有研究进行分类评述。现有研究表明,AGBL5属于M14家族金属羧肽酶,是调控微管蛋白谷氨酰化的核心去谷氨酰化酶,优先去除微管蛋白侧链谷氨酸的分支点,维持谷氨酰化修饰的动态平衡;斑马鱼模型中AGBL5缺失会导致纤毛超谷氨酰化、纤毛运动异常及多系统纤毛病表型,小鼠模型中AGBL5缺失则会引发精子发生障碍与视网膜变性;人类中已报道11种AGBL5错义及移码突变与非综合征型RP相关,证实其为RP的致病基因。现有研究的局限性在于,缺乏人类细胞模型的机制研究,未明确AGBL5缺失导致视网膜变性的具体细胞表型,也未探索针对该致病机制的潜在治疗途径。本文的创新价值在于,首次构建了AGBL5敲除的人类ARPE19细胞模型,系统表征了其超谷氨酰化、纤毛缩短及纤毛形成率降低的细胞表型,并通过实验验证了AGBL5过表达和TTLL5敲低两种策略对上述表型的拯救作用,填补了AGBL5在人类视网膜细胞中功能研究的空白,为AGBL5相关RP提供了全新的潜在治疗方向。
3. 研究思路总结与详细解析
整体研究目标为明确AGBL5在人类视网膜色素上皮细胞中的功能,解析AGBL5缺失的致病机制,并探索潜在的表型拯救策略;核心科学问题是AGBL5缺失如何通过微管蛋白修饰异常导致纤毛缺陷,以及如何逆转该病理表型;技术路线遵循“细胞模型构建→表型系统表征→多策略表型拯救→分子机制探索”的闭环逻辑。
3.1 AGBL5敲除细胞系构建与表型表征
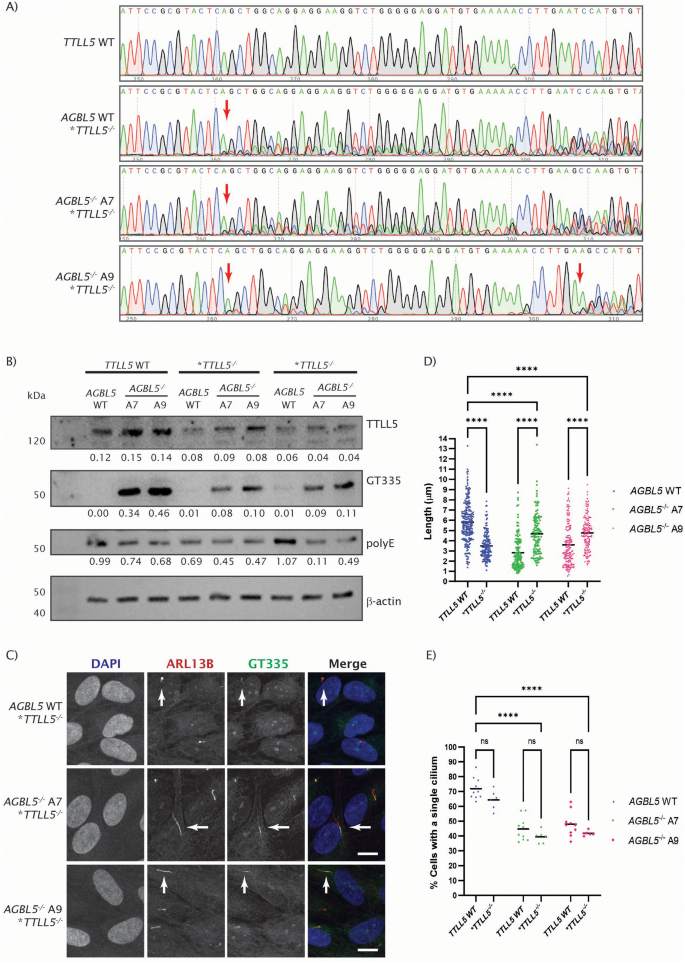
实验目的是构建AGBL5功能缺失的人类视网膜色素上皮细胞模型,明确其病理表型特征。方法细节:采用CRISPR/Cas9技术靶向AGBL5外显子3(两种转录本共有外显子)构建ARPE19敲除克隆,通过Sanger测序验证基因编辑位点,Western blot检测AGBL5蛋白表达水平,RNA-seq分析转录本剪接变化;通过Western blot检测微管蛋白谷氨酰化(GT335抗体标记分支点谷氨酰化、PolyE抗体标记多聚谷氨酰化)水平,免疫荧光结合共聚焦显微镜、Hyvolution超高分辨率成像检测纤毛长度与纤毛形成率。结果解读:测序结果显示敲除克隆存在1bp缺失,引发异常剪接产生截短型AGBL5蛋白;Western blot显示敲除细胞中AGBL5蛋白水平较野生型降低约40%(n=9,P<0.05),GT335标记的谷氨酰化水平显著升高,PolyE标记的多聚谷氨酰化水平降低;免疫荧光结果显示,AGBL5敲除细胞的纤毛长度较野生型缩短51.4%(克隆A7)和38.3%(克隆A9,n=多视野,P<0.0001),纤毛形成率从野生型的70.2%降至40.1%(克隆A7)和46.94%(克隆A9,n=多视野,P<0.0001)。
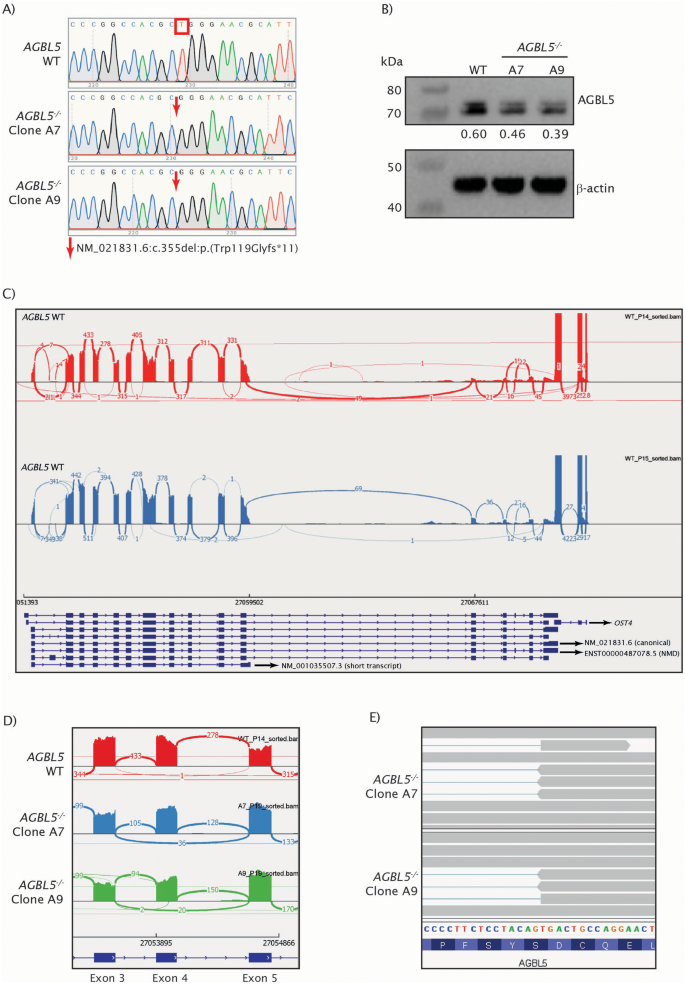
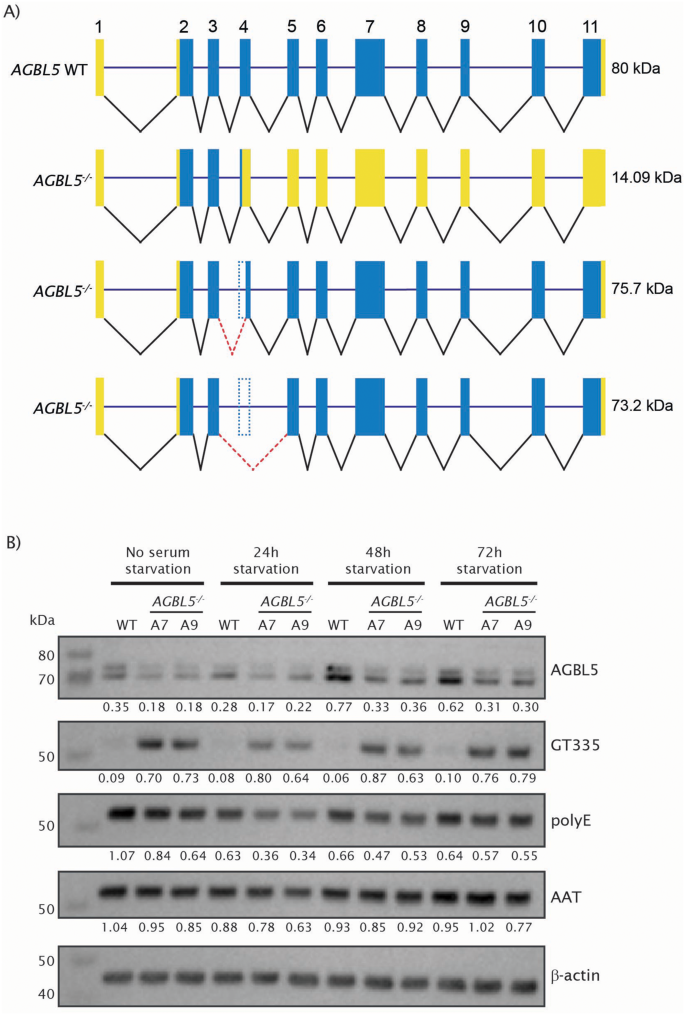
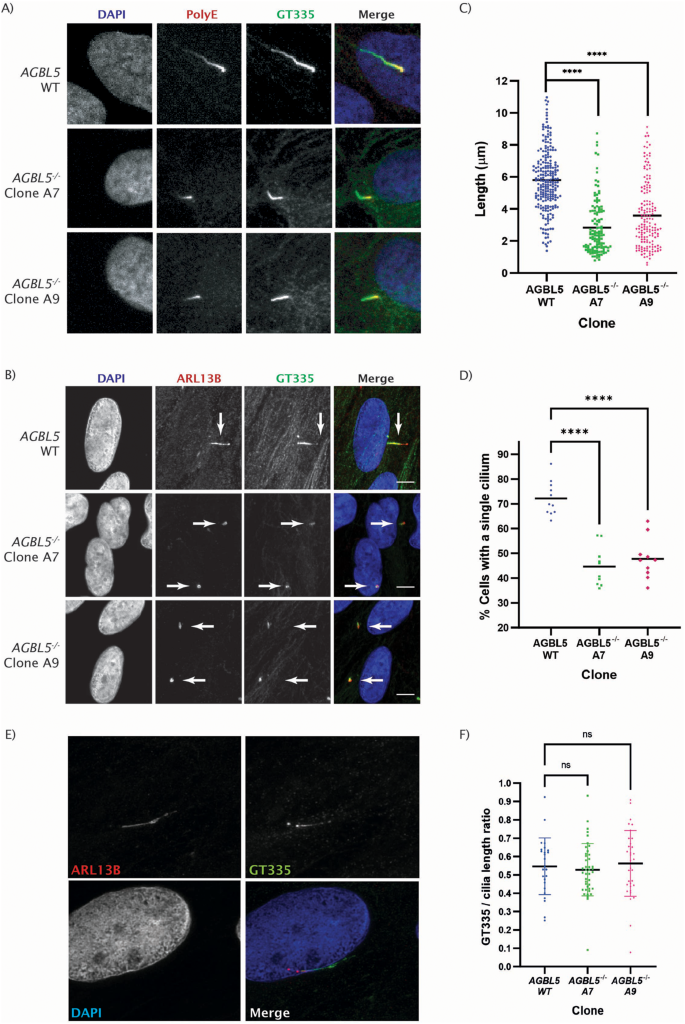
实验所用关键产品:CRISPR/Cas9系统(Synthego)、GT335抗体(Adipogen AG-20B-0020)、ARL13B抗体(Proteintech 17711-1-AP)、Leica SP5共聚焦显微镜、Orbitrap Fusion Lumos质谱仪(Thermo Scientific)。
3.2 AGBL5过表达介导的表型拯救
实验目的是验证AGBL5功能恢复是否能逆转敲除细胞的病理表型。方法细节:将AGBL5-eGFP表达载体转染至AGBL5敲除细胞,同时转染pmaxGFP作为对照,血清饥饿72小时诱导纤毛形成;通过免疫荧光检测纤毛长度与纤毛形成率,Western blot检测不同时间点(24h、48h、72h)的谷氨酰化水平。结果解读:转染AGBL5-eGFP后,敲除细胞的纤毛长度恢复至野生型水平,纤毛形成率从敲除细胞的低水平提升至与野生型无统计学差异(n=多视野,P>0.05);Western blot显示,转染后24h AGBL5-eGFP表达量最高,GT335标记的谷氨酰化水平显著降低至野生型水平,PolyE标记的多聚谷氨酰化水平也同步降低,且血清饥饿细胞中AGBL5-eGFP表达量下降更快。
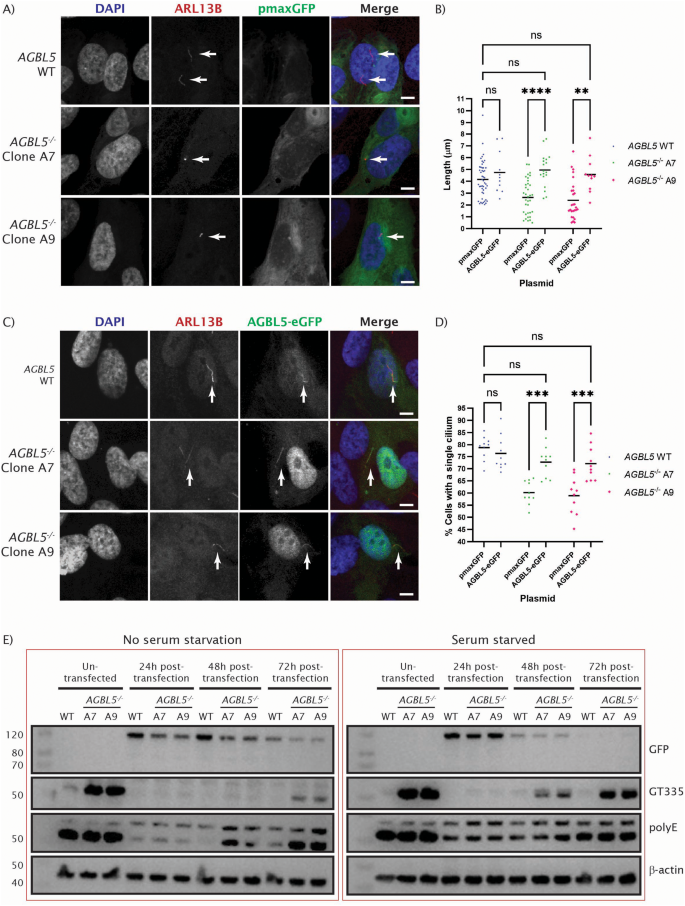
实验所用关键产品:AGBL5-eGFP载体(GenScript)、Lipofectamine RNAiMAX转染试剂(ThermoFisher)、iBright 15000成像系统(ThermoFisher)。
3.3 TTLL5敲低介导的表型拯救
实验目的是探索除AGBL5过表达外的其他潜在表型拯救策略。方法细节:首先通过RNA-seq差异基因分析筛选到RET作为潜在靶点,构建RET敲除细胞池但未观察到表型变化;随后选择谷氨酰化起始酶TTLL5,分别通过CRISPR/Cas9构建TTLL5敲低细胞池,以及siRNA(ON-TARGETplus SMARTpool)敲低TTLL5,检测纤毛表型与谷氨酰化水平。结果解读:CRISPR敲低TTLL5后,AGBL5敲除细胞的纤毛长度部分恢复(n=多视野,P<0.0001),GT335标记的谷氨酰化水平显著降低;siRNA敲低TTLL5后,不仅纤毛长度恢复至接近野生型水平,纤毛形成率也从敲除细胞的低水平提升至接近野生型(n=多视野,P<0.0001),谷氨酰化水平同步降低,且该效果在120h处理后更为显著。
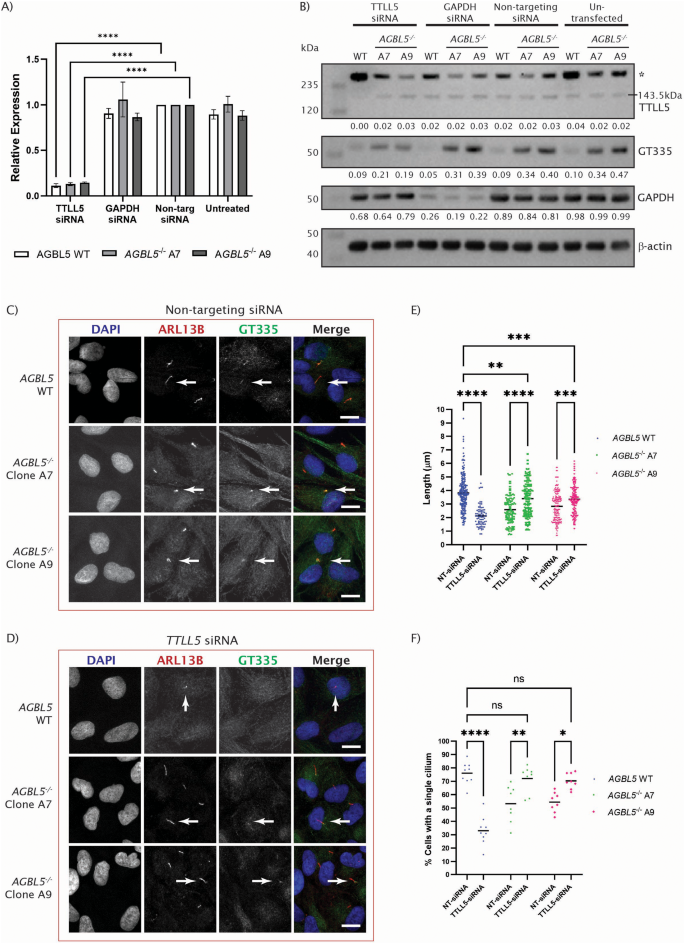
实验所用关键产品:TTLL5 siRNA(Dharmacon ON-TARGETplus SMARTpool)、CRISPR/Cas9系统(Synthego)、StepOnePlus实时荧光定量PCR系统(ThermoFisher)。
3.4 AGBL5相互作用蛋白鉴定
实验目的是探索AGBL5调控纤毛的分子机制,鉴定其潜在的相互作用蛋白。方法细节:转染AGBL5-eGFP至野生型ARPE19细胞,通过GFP-Trap琼脂糖珠沉淀蛋白复合物,采用液相色谱-串联质谱(LC-MS/MS)分析鉴定相互作用蛋白,通过免疫共沉淀验证候选蛋白的相互作用。结果解读:质谱分析筛选到64个与AGBL5显著相互作用的蛋白(log2FC>3,P<0.05),其中包括纤毛相关蛋白EXOC4和USP9X;免疫共沉淀实验显示,内源性AGBL5与EXOC4、USP9X存在特异性相互作用,提示这些蛋白可能参与AGBL5调控纤毛形成与稳定性的分子过程。
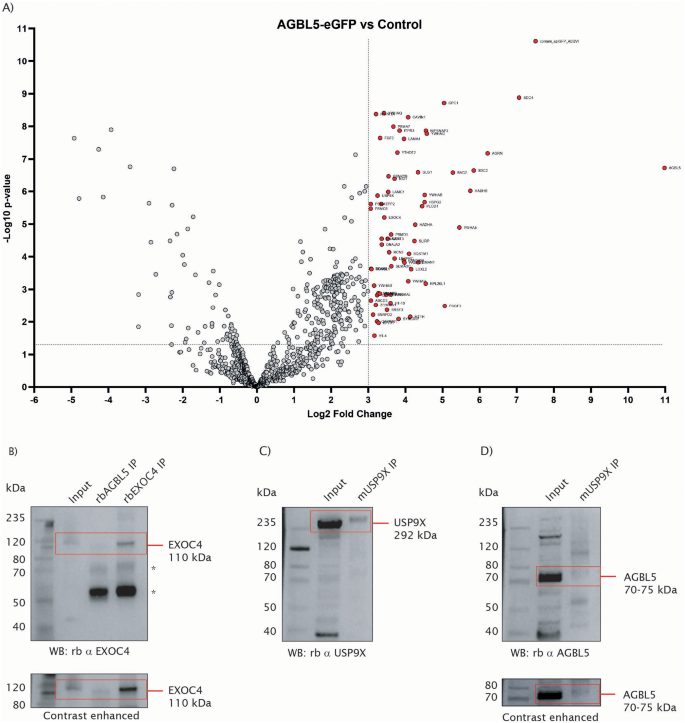
实验所用关键产品:GFP-Trap agarose beads(ChromoTek)、Orbitrap Fusion Lumos质谱仪(Thermo Scientific)、Protein G Plus/Protein A琼脂糖珠(Sigma)。
4. Biomarker研究及发现成果
Biomarker定位:AGBL5作为非综合征型RP的致病基因,其功能缺失导致的微管蛋白超谷氨酰化是疾病相关的分子标志物;TTLL5作为微管蛋白谷氨酰化的起始酶,是AGBL5缺失表型的潜在治疗靶点。研究过程详述:AGBL5的致病突变通过全外显子测序在RP患者中被鉴定,本文在ARPE19细胞模型中验证了AGBL5缺失会导致GT335标记的超谷氨酰化(蛋白水平较野生型升高约2倍,n=3,P<0.01),同时伴随纤毛缩短、纤毛形成率降低的细胞表型;针对TTLL5的干预实验显示,CRISPR敲低和siRNA敲低均能显著降低AGBL5敲除细胞的GT335标记水平(降低约40%-50%,n=3,P<0.01),其中siRNA敲低还能使纤毛形成率从敲除细胞的40%左右提升至65%以上(n=多视野,P<0.0001)。核心成果提炼:AGBL5功能缺失通过破坏微管蛋白谷氨酰化的动态平衡,导致纤毛结构与功能异常,是AGBL5相关RP的核心致病机制;首次证明AGBL5过表达和TTLL5敲低两种策略可逆转AGBL5缺失的病理表型,为AGBL5相关RP提供了基因治疗和反义寡核苷酸治疗的潜在方向;鉴定的EXOC4和USP9X相互作用蛋白,为AGBL5调控纤毛的分子机制研究提供了新的线索。